
Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
La alcaptonuria es una anomalía enzimática congénita
Médico experto del artículo.

Último revisado: 04.07.2025
Uno de los trastornos metabólicos muy raros, la alcaptonuria, se refiere a anomalías congénitas en el metabolismo del aminoácido tirosina.
Este síndrome también puede llamarse deficiencia de homogentisato oxidasa, homogentisinuria, ocronosis hereditaria o enfermedad de la orina negra.[ 1 ]
Epidemiología
Según las estadísticas, no se dan más de nueve casos de alcaptonuria por cada millón de personas. En la mayoría de los países europeos, se da un caso por cada 100.000 a 250.000 nacidos vivos.
Entre los países europeos, la excepción es Eslovaquia (especialmente la relativamente pequeña región noroccidental), donde la prevalencia de alcaptonuria es de un caso por cada 19.000 recién nacidos. Esto probablemente se deba a que, entre las familias romaníes eslovacas que viven allí, el nivel de endogamia (matrimonio entre primos) es el más alto de Europa: entre el 10 % y el 14 %. [ 2 ]
Causas alcaptonuria
Se han establecido las causas exactas de la alcaptonuria, un trastorno congénito del catabolismo (degradación metabólica) del α-aminoácido aromático (homocíclico) tirosina. Este tipo de trastorno metabólico es consecuencia de mutaciones homocigóticas o heterocigóticas compuestas de uno de los miles de genes del cromosoma 3, concretamente, el gen HGD, ubicado en el locus 3q21-q23 del brazo largo del cromosoma. Este gen codifica las secuencias de nucleótidos de la enzima hepática homogentisato-1,2-dioxigenasa [ 3 ] (también llamada oxidasa del ácido homogentísico u homogentisato oxidasa), una metaloproteína que contiene hierro, necesaria para una de las etapas de la degradación de la tirosina en el organismo. [ 4 ], [ 5 ]
Así, la alcaptonuria es un defecto de la enzima homogentisato-1,2-dioxigenasa, o más precisamente, el resultado de su deficiencia o ausencia completa determinada genéticamente. [ 6 ]
Al ser una deficiencia enzimática congénita, la alcaptonuria se hereda como un rasgo autosómico recesivo, es decir, para que la alcaptonuria se presente en los hijos, ambos padres deben tener un gen modificado para la enzima, ya que cada uno de ellos transmite al hijo sólo una copia del gen de las dos disponibles.
Según los últimos datos, existen más de doscientas variantes de modificación del gen HGD y las mutaciones sin sentido, la translocación y el empalme son los más frecuentemente observados.
Factores de riesgo
El único factor de riesgo para desarrollar esta enzimopatía congénita es su presencia en la historia familiar y la herencia de dos copias modificadas del gen HGD, si los padres no presentan alcaptonuria (el riesgo de transmitir la anomalía es del 25%), o uno de los padres tiene este trastorno. [ 7 ]
Patogenesia
La tirosina desempeña un papel clave en la síntesis de proteínas, la producción de cromoproteínas (el pigmento de la piel, melanina), así como de hormonas tiroideas y neurotransmisores de catecolaminas.
El mecanismo para regular la cantidad de tirosina en las células es muy complejo, y el organismo normaliza su exceso descomponiéndola. El catabolismo de la tirosina, como el de todos los aminoácidos aromáticos, es multietapa y se produce en varias etapas. Cada etapa de la degradación metabólica de la tirosina se produce con la participación de una enzima específica y la formación de un compuesto intermedio.
Así, primero el aminoácido se descompone en para-hidroxifenilpiruvato, que se convierte en alcaptona (ácido 2,5-dihidroxifenilacético u homogentísico). Posteriormente, la alcaptona debería transformarse en ácido maleacético, pero esto no ocurre. [ 8 ]
Y la patogenia de la alcaptonuria consiste en el cese de las reacciones bioquímicas del catabolismo de la tirosina en la etapa de formación del ácido homogentísico: simplemente no se necesita ninguna enzima para descomponerlo: la homogentisato oxidasa.
El ácido homogentísico no es aprovechado por el organismo y puede acumularse al excretarse por los riñones. Además, se oxida a benzoquinoacetato (ácido benzoquinonaacético), el cual, al unirse a las moléculas de los tejidos y fluidos corporales, forma compuestos biopoliméricos de color similar a la melanina.
La acumulación de estos productos intermedios en el tejido conduce a una alteración de la estructura del colágeno del tejido del cartílago, lo que reduce su elasticidad, con la aparición de muchos signos clínicos de alcaptonuria y el desarrollo de complicaciones.
Síntomas alcaptonuria
La alcaptonuria en recién nacidos y lactantes se caracteriza por el oscurecimiento de la orina. Al exponerse al aire, la orina en pañales y ropa interior se vuelve marrón oscuro; esto se debe a la acumulación y liberación de ácido homogentísico, que se oxida a benzoquinoacetato. [ 9 ]
En ausencia de otros síntomas, la alcaptonuria en niños pequeños a menudo no se detecta a tiempo, ya que la orina puede oscurecerse después de varias horas de orinar. Según algunos datos, solo una quinta parte de los niños menores de 12 meses que nacen con esta deficiencia enzimática se diagnostican en el ámbito clínico. Por lo tanto, es fundamental que los padres presten atención al cuidado de sus bebés.
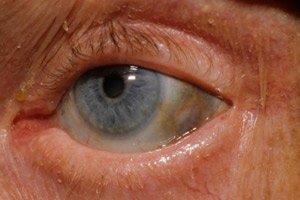
Además, los primeros signos incluyen la pigmentación (color gris azulado) de la esclerótica de los ojos y los cartílagos de las orejas y la nariz, lo que a menudo se denomina ocronosis.[ 10 ]
Con el tiempo aparecen otros síntomas:
- pigmentación severa de la piel en los pómulos, axilas y genitales;
- manchas en la ropa al entrar en contacto con zonas sudorosas del cuerpo;
- ataques de debilidad general;
- voz ronca.
Debe tenerse en cuenta que la alcaptonuria y la ocronosis, como se señaló anteriormente, son nombres sinónimos para el mismo trastorno del catabolismo de la tirosina.
Enfermedad de la orina con olor a jarabe de arce y alcaptonuria. La enfermedad congénita de la orina con olor a jarabe de arce o leucinosis es también un trastorno metabólico, tiene el mismo patrón de herencia e incluso las mutaciones ocurren en el mismo cromosoma, pero afectan al gen que codifica el complejo enzimático de la α-cetoácido deshidrogenasa de cadena ramificada. Debido a esto, el cuerpo no puede descomponer ciertos componentes de las proteínas, en particular, los aminoácidos leucina, isoleucina y valina. Con esta enfermedad, la orina (y el cerumen) tienen un olor dulce; además, el cuadro clínico de este tipo de acidemia orgánica incluye hipopigmentación, fluctuaciones en la presión arterial, convulsiones, vómitos y diarrea, un descenso en los niveles de glucosa en sangre, cetoacidosis, alucinaciones, etc. La tasa de mortalidad en niños es bastante alta; en adultos, sin tratamiento, puede ocurrir coma y muerte debido a edema cerebral.
El albinismo y la alcaptonuria se unen únicamente por la tirosina. El albinismo, incluido el oculocutáneo, se debe a mutaciones genéticas que afectan la producción del pigmento melanina. Se observan alteraciones congénitas en el gen TYR del cromosoma 11 (11q14.3), que codifica la tirosinasa, una enzima del melanosoma que contiene cobre, necesaria para la formación del pigmento cutáneo a partir de los productos del metabolismo de la tirosina. Esta enfermedad es mucho más común que la alcaptonuria.
Complicaciones y consecuencias
Las consecuencias y complicaciones de la alcaptonuria, causadas por la acción de los metabolitos intermedios de la tirosina (ácidos homogentísico y benzoquinonaacético), aparecen debido a la deposición de polímeros pigmentados reactivos, la destrucción de las fibrillas de colágeno y el deterioro del estado del cartílago (con una disminución de su resistencia al estrés mecánico).
Con el paso de los años, en la edad adulta, se desarrollan artritis degenerativa y osteoartritis de las articulaciones grandes (cadera, sacroilíaca y rodilla); los espacios intervertebrales se estrechan (especialmente en la columna lumbar y torácica), con calcificación y formación de osteofitos; la densidad del tejido de las placas óseas subcondrales disminuye y los huesos subyacentes pueden sufrir una remodelación patológica con la formación de crecimientos y deformación. [ 11 ]
Se pueden observar daños en las válvulas cardíacas (aórtica y mitral) y en las arterias coronarias, con signos de enfermedad coronaria, así como la formación de cálculos en los riñones y la próstata, debido a la misma calcificación. [ 12 ], [ 13 ]
Diagnostico alcaptonuria
Normalmente, el diagnóstico de los trastornos metabólicos congénitos se basa en el estudio de los fluidos biológicos del cuerpo.
¿Qué pruebas y reacciones se utilizan para diagnosticar la alcaptonuria? Se requieren análisis de orina para detectar ácido homogentísico y determinar su nivel (normal: 20-30 mg al día, elevado: 3-8 g). Se examina una muestra de orina mediante cromatografía de gases o espectrometría de masas, utilizando cromatografía líquida; es posible realizar una prueba de detección de cloruro de hierro en la orina. [ 14 ]
También existe un método para el diagnóstico rápido: la determinación de alcaptona en manchas de orina seca sobre papel (por la intensidad del color).
Para aclarar el diagnóstico, el diagnóstico instrumental (radiografía) implica identificar signos radiológicos de osteoartritis y otras patologías articulares en los pacientes.
El diagnóstico se confirma mediante métodos genéticos moleculares de diagnóstico de enfermedades hereditarias, como pruebas genéticas y secuenciación de ADN. [ 15 ]
Diagnóstico diferencial
El diagnóstico diferencial incluye hemocromatosis e insuficiencia hepática aguda del recién nacido, melaninuria, porfiria intermitente aguda, linfohistiocitosis hemofagocítica, patología mitocondrial primaria, artritis reumatoide y espondilitis anquilosante.
¿A quién contactar?
Tratamiento alcaptonuria
El principal tratamiento para la alcaptonuria es la administración oral de grandes dosis (al menos 1000 mg al día) de ácido ascórbico. En niños, esto aumenta la excreción urinaria de ácido homogentísico, y en adultos, reduce el contenido urinario de su derivado, el ácido benzoquinonaacético, y retarda su unión a las estructuras del tejido conectivo de las articulaciones y al colágeno. [ 16 ]
Clínicas de Europa Occidental están probando el fármaco nitisinona (Orfalin), un fármaco del grupo de metabolitos que inhibe la segunda etapa del catabolismo de la tirosina: la transformación del para-hidroxifenilpiruvato en ácido homogentísico. Sin embargo, el uso de este fármaco provoca la acumulación de tirosina y puede causar efectos secundarios graves, como opacidad corneal y fotofobia, hemorragias nasales y estomacales, insuficiencia hepática, alteraciones sanguíneas, etc. No obstante, en Estados Unidos, la nitisinona está aprobada por la FDA para el tratamiento de la tirosinemia tipo I. [17 ], [ 18 ]
Por ello, para los problemas articulares causados por la alcaptonuria se realiza un tratamiento de fisioterapia (terapia de ejercicios para aumentar la fuerza muscular y mejorar la movilidad articular, balneoterapia y terapia peloide para limitar el dolor).
Aunque la tirosina no sólo es aportada por los alimentos sino que también se produce en el organismo, a los pacientes con alcaptonuria se les recomienda seguir una dieta baja en proteínas y limitar el consumo de alimentos ricos en tirosina, principalmente carne de res y de cerdo, productos lácteos (especialmente quesos), legumbres, frutos secos y semillas.
Prevención
La prevención de las mutaciones genéticas es imposible, pero para prevenir el nacimiento de niños con alto riesgo de trastornos congénitos, existe el asesoramiento genético médico, que es necesario antes de un embarazo planificado para aquellas parejas cuyos antecedentes familiares incluyen enfermedades hereditarias. [ 19 ]
Pronóstico
Los desenlaces mortales por alcaptonuria son muy poco frecuentes, y la muerte puede deberse a complicaciones graves que afectan al corazón y los riñones. Por lo tanto, la esperanza de vida general de las personas con alcaptonuria es buena.
Pero la calidad de vida se ve reducida debido al intenso dolor en las articulaciones o la columna con importante limitación de la movilidad, muchas veces progresiva.