
Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
Epilepsia criptogénica con crisis en adultos
Médico experto del artículo.

Último revisado: 04.07.2025
Según la clasificación internacional vigente hasta el año pasado, se distinguía entre la epilepsia sintomática o secundaria, causada por daño a las estructuras cerebrales, la idiopática, la primaria (una enfermedad independiente, presumiblemente hereditaria) y la criptogénica. Esta última opción implica que los diagnósticos modernos no han establecido ninguna causa para las crisis epilépticas periódicas, ni se ha rastreado la predisposición hereditaria. El concepto mismo de "criptogénico" se traduce del griego como "origen desconocido" (kryptos: secreto, secreto, genos: generado).
La ciencia no se detiene y, quizás, pronto se determine el origen de las crisis epilépticas periódicas de etiología desconocida. Los expertos sugieren que la epilepsia criptogénica es una enfermedad sintomática secundaria, cuya génesis no puede determinarse con el nivel actual de diagnóstico.
 [
[ Epidemiología
La epilepsia y los síndromes epilépticos son patologías neurológicas muy comunes, que a menudo conllevan consecuencias graves. Las crisis epilépticas pueden presentarse en personas de cualquier género y edad. Se estima que aproximadamente el 5% de la población mundial ha sufrido al menos una crisis a lo largo de su vida.
Cada año, se diagnostica epilepsia o síndrome epiléptico en un promedio de 30 a 50 personas por cada 100 mil habitantes del planeta. Las crisis epilépticas se presentan con mayor frecuencia en bebés (entre 100 y 233 casos por cada 100 mil personas). El pico de manifestación se produce en el período perinatal, tras lo cual la tasa de incidencia disminuye casi a la mitad. Las tasas más bajas se observan en personas de 25 a 55 años, con alrededor de 20 a 30 casos por cada 100 mil personas. Posteriormente, la probabilidad de sufrir crisis epilépticas aumenta y, a partir de los 70 años, la tasa de incidencia es de 150 casos o más por cada 100 mil personas.
Las causas de la epilepsia se establecen en aproximadamente el 40% de los casos, por lo que una enfermedad de etiología desconocida no es infrecuente. Los espasmos infantiles (síndrome de West), una epilepsia criptogénica, se diagnostican en niños de cuatro a seis meses de edad; en promedio, se diagnostica un niño por cada 3200 bebés.
Causas epilepsia criptogénica
La base para el diagnóstico de la epilepsia son las convulsiones periódicas, cuya causa es una descarga eléctrica anormalmente fuerte, que es el resultado de la sincronización de la actividad de las células cerebrales en todos los rangos de frecuencia, que se expresa externamente en la aparición de síntomas sensoriomotores, neurológicos y mentales.
Para que se produzca una crisis epiléptica, es necesaria la presencia de las llamadas neuronas epilépticas, que se caracterizan por la inestabilidad del potencial de reposo (la diferencia de potenciales entre una célula no excitada en las caras interna y externa de su membrana). Como resultado, el potencial de acción de una neurona epiléptica excitada presenta una amplitud, duración y frecuencia significativamente superiores a las normales, lo que conduce al desarrollo de una crisis epiléptica. Se cree que las crisis se producen en personas con predisposición hereditaria a estos cambios, es decir, en grupos de neuronas epilépticas capaces de sincronizar su actividad. Los focos epilépticos también se forman en áreas del cerebro con estructura alterada debido a lesiones, infecciones, intoxicaciones y desarrollo de tumores.
Así pues, en pacientes con diagnóstico de epilepsia criptogénica, los métodos modernos de neuroimagen no detectan anomalías en la estructura de la masa cerebral y no hay antecedentes familiares de epilepsia. Sin embargo, los pacientes experimentan convulsiones epilépticas de diversos tipos con bastante frecuencia, difíciles de tratar (posiblemente precisamente porque su causa es desconocida).
Por consiguiente, los factores de riesgo conocidos para la aparición de crisis epilépticas (genética, alteración de la estructura del cerebro, procesos metabólicos en sus tejidos, consecuencias de traumatismos craneoencefálicos o procesos infecciosos) no se detectan durante los exámenes y encuestas.
Según la nueva clasificación de las epilepsias de 2017, se distinguen seis categorías etiológicas de la enfermedad. En lugar de sintomática, ahora se recomienda determinar el tipo de epilepsia según la causa establecida: estructural, infecciosa, metabólica, inmunitaria o una combinación de ellas. La epilepsia idiopática presupone la presencia de una predisposición hereditaria y ahora se denomina genética. El término "criptogénico" se ha sustituido por "factor etiológico desconocido", lo que ha aclarado el significado de la expresión, pero no ha cambiado.
La patogenia de la epilepsia es presumiblemente la siguiente: formación de un foco epiléptico, es decir, una comunidad de neuronas con electrogénesis alterada → creación de sistemas epilépticos en el cerebro (con una liberación excesiva de mediadores excitatorios, se lanza una “cascada de glutamato”, que afecta a todas las neuronas nuevas y contribuye a la formación de nuevos focos de epileptogénesis) → formación de conexiones interneuronales patológicas → se produce la generalización de la epilepsia.
La principal hipótesis sobre el mecanismo de desarrollo de la epilepsia es que el proceso patológico se desencadena por una alteración del equilibrio entre los neurotransmisores excitatorios (glutamato, aspartato) y los responsables de los procesos de inhibición (ácido γ-aminobutírico, taurina, glicina, noradrenalina, dopamina, serotonina). Se desconoce qué altera exactamente este equilibrio en nuestro caso. Sin embargo, como resultado, las membranas celulares de las neuronas se ven afectadas, la cinética de los flujos iónicos se altera: las bombas iónicas se inactivan y, a la inversa, los canales iónicos se activan, alterándose la concentración intracelular de iones de potasio, sodio y cloro con carga positiva. El intercambio iónico patológico a través de membranas desestructuradas provoca cambios en el flujo sanguíneo cerebral. La disfunción de los receptores de glutamato y la producción de autoanticuerpos contra ellos causan convulsiones epilépticas. Las descargas neuronales excesivamente intensas y periódicas que se manifiestan en forma de convulsiones epilépticas provocan alteraciones profundas de los procesos metabólicos en las células de la sustancia cerebral y provocan el desarrollo de la siguiente convulsión.
La especificidad de este proceso reside en la agresividad de las neuronas del foco epiléptico en relación con las áreas cerebrales aún inalteradas, lo que les permite subyugar nuevas áreas. La creación de sistemas epilépticos ocurre durante el proceso de formación de relaciones patológicas entre el foco epiléptico y los componentes estructurales del cerebro capaces de activar el mecanismo de desarrollo de la epilepsia. Dichas estructuras incluyen: el tálamo, el sistema límbico y la formación reticular de la parte media del tronco encefálico. Por el contrario, las relaciones que surgen con el cerebelo, el núcleo caudado de la subcorteza y la corteza orbital anterior ralentizan el desarrollo de la epilepsia.
Durante el desarrollo de la enfermedad, se forma un sistema patológico cerrado: el cerebro epiléptico. Su formación culmina con un trastorno del metabolismo celular y la interacción de neurotransmisores, la circulación cerebral, una creciente atrofia de los tejidos y vasos cerebrales y la activación de procesos autoinmunes cerebrales específicos.
Síntomas epilepsia criptogénica
La principal manifestación clínica de esta enfermedad es una crisis epiléptica. Se sospecha epilepsia cuando el paciente ha presentado al menos dos crisis epilépticas reflejas (no provocadas), cuyas manifestaciones son muy diversas. Por ejemplo, las crisis epilépticas similares a las causadas por altas temperaturas que no se presentan en un estado normal no se consideran epilepsia.
Los pacientes con epilepsia criptogénica pueden experimentar convulsiones de distintos tipos y con bastante frecuencia.
Los primeros signos del desarrollo de la enfermedad (antes de la aparición de convulsiones epilépticas completas) pueden pasar desapercibidos. El grupo de riesgo incluye a las personas que sufrieron convulsiones febriles en la primera infancia, con una mayor predisposición a las convulsiones. En el período prodrómico, pueden observarse trastornos del sueño, mayor irritabilidad y labilidad emocional.
Además, los ataques no siempre se presentan en la forma generalizada clásica con caídas, convulsiones y pérdida de conocimiento.
A veces, los únicos signos iniciales son trastornos del habla: el paciente está consciente pero no habla ni responde preguntas, o desmayos breves y periódicos. Esto no dura mucho, solo un par de minutos, por lo que pasa desapercibido.
Las convulsiones focales o parciales (locales o limitadas) simples son más frecuentes, y sus manifestaciones dependen de la localización del foco epiléptico. El paciente no pierde el conocimiento durante el paroxismo.
Durante una convulsión motora simple, pueden observarse tics, espasmos en las extremidades, calambres musculares y movimientos de rotación del torso y la cabeza. El paciente puede emitir sonidos inarticulados o permanecer en silencio, sin responder preguntas, chasqueando los labios, lamiéndose los labios y haciendo movimientos de masticación.
Las convulsiones sensitivas simples se caracterizan por parestesias (entumecimiento de varias partes del cuerpo, sensaciones inusuales de gusto o de olor, generalmente desagradables); alteraciones visuales (destellos de luz, una cuadrícula, manchas delante de los ojos, visión de túnel).
Los paroxismos vegetativos se manifiestan por palidez repentina o hiperemia de la piel, aumento del ritmo cardíaco, saltos en la presión arterial, constricción o dilatación de las pupilas, molestias en la zona del estómago hasta dolor y vómitos.
Las convulsiones mentales se manifiestan por desrealización/despersonalización y ataques de pánico. Por lo general, son precursoras de convulsiones focales complejas, que ya se acompañan de alteración de la consciencia. El paciente comprende que está sufriendo una convulsión, pero no puede buscar ayuda. Los sucesos ocurridos durante la convulsión se borran de su memoria. Las funciones cognitivas de la persona se ven afectadas: se produce una sensación de irrealidad ante lo que está sucediendo y aparecen nuevos cambios en su interior.
Las convulsiones focales con generalización posterior comienzan como simples (complejas) y se transforman en paroxismos tónico-clónicos generalizados. Duran unos tres minutos y culminan en sueño profundo.
Las convulsiones generalizadas se presentan de forma más grave y se dividen en:
- tónico-clónico, que se presenta en la siguiente secuencia: el paciente pierde el conocimiento, se cae, su cuerpo se dobla y se estira en un arco, comienzan contracciones convulsivas de los músculos de todo el cuerpo; los ojos del paciente giran hacia atrás, sus pupilas se dilatan en este momento; el paciente grita, se pone azul como resultado de dejar de respirar durante varios segundos, se observa hipersalivación espumosa (la espuma puede adquirir un tinte rosado debido a la presencia de sangre en ella, lo que indica morderse la lengua o la mejilla); a veces se produce un vaciado involuntario de la vejiga;
- Las convulsiones mioclónicas se presentan como contracciones intermitentes (rítmicas y arrítmicas) de los músculos durante varios segundos en todo el cuerpo o en ciertas áreas del cuerpo, que parecen aleteo de extremidades, sentadillas, apretar los puños y otros movimientos monótonos; la conciencia, especialmente en las convulsiones focales, se conserva (este tipo se observa con mayor frecuencia en la infancia);
- ausencias - convulsiones no convulsivas con pérdida de conciencia a corto plazo (5-20 segundos), expresadas en el hecho de que una persona se congela con los ojos abiertos e inexpresivos y no reacciona a los estímulos, generalmente no se cae, al volver en sí, continúa la actividad interrumpida y no recuerda la convulsión;
- Las ausencias atípicas se acompañan de caídas, vaciado involuntario de la vejiga, son más duraderas y se presentan en formas graves de la enfermedad, combinadas con retraso mental y otros síntomas de trastornos mentales;
- convulsiones atónicas (acinéticas): el paciente cae bruscamente como resultado de la pérdida del tono muscular (en las epilepsias focales, puede haber atonía de grupos musculares individuales: faciales, caída de la mandíbula inferior, cervicales, el paciente se sienta o se para con la cabeza colgando), la duración de la convulsión no es más de un minuto; la atonía en las ausencias ocurre gradualmente: el paciente se hunde lentamente, en convulsiones atónicas aisladas, cae bruscamente.
En el período posterior a las convulsiones, el paciente está letárgico e inhibido; si no se le molesta, se queda dormido (especialmente después de las convulsiones generalizadas).
Los tipos de epilepsia corresponden a los tipos de convulsiones. Las convulsiones focales (parciales) se desarrollan en un foco epiléptico local, cuando una descarga anormalmente intensa encuentra resistencia en las zonas vecinas y se extingue sin propagarse a otras partes del cerebro. En estos casos, se diagnostica epilepsia focal criptogénica.
El curso clínico de la enfermedad con un foco epiléptico limitado (forma focal) está determinado por su localización.
Con mayor frecuencia, se observa daño en la región temporal. Esta forma de epilepsia es progresiva y las convulsiones suelen ser mixtas, con una duración de varios minutos. La epilepsia temporal criptogénica, fuera de las convulsiones, se manifiesta con cefaleas, mareos constantes y náuseas. Los pacientes con esta forma de localización se quejan de micción frecuente. Antes de la convulsión, sienten un aura premonitoria.
La lesión puede localizarse en el lóbulo frontal del cerebro. Las convulsiones se caracterizan por su brusquedad, sin aura prodrómica. El paciente presenta espasmos craneales, ojos que giran hacia abajo y hacia los lados, y una gesticulación automática y bastante compleja es característica. El paciente puede perder el conocimiento, caerse y presentar espasmos musculares tónico-clónicos en todo el cuerpo. En esta localización, se observa una serie de convulsiones breves, a veces con transición a generalizadas o estado epiléptico. Pueden comenzar no solo durante la vigilia diurna, sino también durante el sueño nocturno. La epilepsia frontal criptogénica, en desarrollo, causa trastornos mentales (pensamiento violento, desrealización) y del sistema nervioso autónomo.
Convulsiones sensoriales (sensación de aire caliente moviéndose sobre la piel, toques ligeros) combinadas con espasmos convulsivos de partes del cuerpo, trastornos del habla y motores, atonía, acompañada de incontinencia urinaria.
La localización del foco epiléptico en la región orbitofrontal se manifiesta por alucinaciones olfativas, hipersalivación, malestar epigástrico, así como trastornos del habla, tos y edema laríngeo.
Si la hiperactividad eléctrica se propaga por todo el cerebro, se desarrolla una convulsión generalizada. En este caso, se diagnostica al paciente con epilepsia generalizada criptogénica. En este caso, las convulsiones se caracterizan por su intensidad, pérdida de consciencia y terminan con un sueño prolongado. Al despertar, los pacientes se quejan de dolores de cabeza, fenómenos visuales, fatiga y sensación de vacío.
También existe un tipo de epilepsia combinada (cuando se producen convulsiones focales y generalizadas) y desconocida.
La epilepsia criptogénica en adultos se considera, con razón, secundaria a un factor etiológico no especificado. Se caracteriza por convulsiones repentinas. Más allá de los síntomas clínicos, los epilépticos presentan inestabilidad mental, temperamento explosivo y tendencia a la agresión. La enfermedad suele comenzar con manifestaciones de alguna forma focal. A medida que progresa, las lesiones se extienden a otras partes del cerebro; la etapa avanzada se caracteriza por degradación personal y desviaciones mentales pronunciadas, y el paciente presenta inadaptación social.
La enfermedad tiene un curso progresivo y los síntomas clínicos de la epilepsia cambian dependiendo del estadio de desarrollo de la epilepsia (el grado de prevalencia del foco epiléptico).
Complicaciones y consecuencias
Incluso en casos leves de epilepsia focal con convulsiones aisladas y poco frecuentes, las fibras nerviosas se dañan. La enfermedad tiene un curso progresivo: una convulsión aumenta la probabilidad de otra y el área de daño cerebral se expande.
Los paroxismos frecuentes y generalizados tienen un efecto destructivo sobre el tejido cerebral y pueden derivar en estado epiléptico con una alta probabilidad de desenlace fatal. También existe riesgo de edema cerebral.
Las complicaciones y consecuencias dependen del grado de daño a las estructuras cerebrales, la gravedad y frecuencia de las convulsiones, las enfermedades concomitantes, la presencia de malos hábitos, la edad, la idoneidad de las tácticas de tratamiento elegidas y las medidas de rehabilitación y la actitud responsable hacia el tratamiento del propio paciente.
A cualquier edad, pueden producirse lesiones de diversa gravedad durante las caídas. La hipersalivación y la tendencia a las arcadas durante una convulsión aumentan el riesgo de que entren sustancias líquidas en el sistema respiratorio y se desarrolle neumonía por aspiración.
En la infancia, el desarrollo mental y físico es inestable. Las capacidades cognitivas suelen verse afectadas.
El estado psicoemocional es inestable: los niños son irritables, caprichosos, a menudo agresivos o apáticos, carecen de autocontrol y se adaptan mal al grupo.
En los adultos, estos riesgos se agravan por las lesiones sufridas al realizar trabajos que requieren mayor atención. Durante las convulsiones, se muerde la lengua o la mejilla.
Las personas con epilepsia tienen un mayor riesgo de desarrollar depresión, trastornos mentales e inadaptación social. Las personas con epilepsia tienen limitaciones en la actividad física y la elección de profesión.
Diagnostico epilepsia criptogénica
En el diagnóstico de la epilepsia se utilizan muchos métodos diferentes para ayudar a diferenciar esta enfermedad de otras patologías neurológicas.
En primer lugar, el médico debe escuchar las quejas del paciente o de sus padres, si se trata de un niño. Se elabora una anamnesis de la enfermedad: detalles de las manifestaciones, las particularidades de la evolución (frecuencia de las convulsiones, desmayos, naturaleza de las convulsiones y otros matices), la duración de la enfermedad y la presencia de enfermedades similares en los familiares del paciente. Este estudio permite determinar el tipo de epilepsia y la localización del foco epiléptico.
Se prescriben análisis de sangre y orina para evaluar el estado general del cuerpo, la presencia de factores como infecciones, intoxicaciones, trastornos bioquímicos y para determinar la presencia de mutaciones genéticas en el paciente.
Se realizan pruebas neuropsicológicas para evaluar las capacidades cognitivas y el estado emocional. El seguimiento periódico permite evaluar el impacto de la enfermedad en el sistema nervioso y la psique, y también ayuda a determinar el tipo de epilepsia.
Sin embargo, en primer lugar, se trata de un diagnóstico instrumental, gracias al cual es posible evaluar la intensidad de la actividad eléctrica de las regiones cerebrales (electroencefalografía), la presencia de malformaciones vasculares, neoplasias, trastornos metabólicos, etc. en sus regiones.
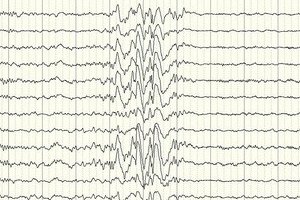
La electroencefalografía (EEG) es el principal método diagnóstico, ya que muestra desviaciones de la norma en la intensidad de las ondas cerebrales incluso fuera de un ataque: aumento de la predisposición a las convulsiones en ciertas áreas o en todo el cerebro. El patrón EEG de la epilepsia parcial criptogénica es una actividad punta-onda o de ondas lentas sostenidas en ciertas partes del cerebro. Mediante este estudio, el tipo de epilepsia se puede determinar con base en la especificidad del electroencefalograma. Por ejemplo, el síndrome de West se caracteriza por ondas lentas arrítmicas irregulares, prácticamente desincronizadas, con una amplitud anormalmente alta y descargas de punta. En la mayoría de los casos de síndrome de Lennox-Gastaut, el electroencefalograma durante la vigilia revela una actividad punta-onda lenta generalizada irregular con una frecuencia de 1,5-2,5 Hz, a menudo con asimetría de amplitud. Durante el descanso nocturno, este síndrome se caracteriza por el registro de descargas rítmicas rápidas con una frecuencia de aproximadamente 10 Hz.
En el caso de la epilepsia criptogénica, esta es la única manera de confirmar su presencia. Aunque hay casos en los que, incluso inmediatamente después de una convulsión, el EEG no registra cambios en la forma de las ondas cerebrales. Esto podría indicar que se producen cambios en la actividad eléctrica en las estructuras profundas del cerebro. Los cambios en el EEG también pueden presentarse en pacientes sin epilepsia.
Se utilizan necesariamente métodos modernos de neurovisualización: computador, resonancia magnética y tomografía por emisión de positrones. Este diagnóstico instrumental permite evaluar cambios en la estructura del cerebro debido a lesiones, anomalías congénitas, enfermedades e intoxicaciones, así como detectar neoplasias, etc. La tomografía por emisión de positrones, también llamada resonancia magnética funcional, ayuda a identificar trastornos tanto estructurales como funcionales.
Se pueden detectar focos más profundos de actividad eléctrica anormal mediante tomografía computarizada por emisión de fotón único, y la espectroscopia de resonancia puede detectar alteraciones en los procesos bioquímicos del tejido cerebral.
Un método de diagnóstico experimental y poco extendido es la magnetoencefalografía, que registra las ondas magnéticas emitidas por las neuronas cerebrales. Permite estudiar las estructuras cerebrales más profundas, inaccesibles a la electroencefalografía.
Diagnóstico diferencial
El diagnóstico diferencial se realiza tras realizar estudios exhaustivos. El diagnóstico de epilepsia criptogénica se realiza descartando otros tipos y causas de crisis epilépticas identificados durante el proceso diagnóstico, así como la predisposición hereditaria.
No todas las instituciones médicas tienen el mismo potencial diagnóstico, por lo que un diagnóstico de este tipo requiere una mayor investigación diagnóstica a un nivel superior.
Tratamiento epilepsia criptogénica
No existe un método único para tratar la epilepsia, sin embargo se han desarrollado estándares claros que se siguen para mejorar la calidad del tratamiento y la vida de los pacientes.
Prevención
Dado que no se han establecido las causas de este tipo particular de epilepsia, las medidas preventivas tienen un enfoque general. Un estilo de vida saludable (sin malos hábitos, buena nutrición y actividad física) fortalece el sistema inmunitario y previene el desarrollo de infecciones.
Prestar mucha atención a su salud, el examen y tratamiento oportuno de enfermedades y lesiones también aumenta la probabilidad de evitar esta enfermedad.
Pronóstico
La epilepsia criptogénica se manifiesta a cualquier edad y no presenta un complejo sintomático específico, sino que se manifiesta de forma muy diversa: pueden presentarse diferentes tipos de convulsiones y síndromes. Hasta la fecha, no existe un único método para la cura completa de la epilepsia, pero el tratamiento antiepiléptico es eficaz en el 60-80 % de los casos de todo tipo de enfermedades.
En promedio, la enfermedad dura 10 años, tras los cuales las convulsiones pueden cesar. Sin embargo, entre el 20 % y el 40 % de los pacientes padecen epilepsia de por vida. Aproximadamente un tercio de los pacientes con cualquier tipo de epilepsia fallecen por causas asociadas.
Por ejemplo, las formas criptogénicas del síndrome de West tienen un pronóstico desfavorable. En la mayoría de los casos, evolucionan al síndrome de Lennox-Gastaut, cuyas formas leves se pueden controlar con fármacos, mientras que las formas generalizadas, con convulsiones frecuentes y graves, pueden persistir de por vida y estar acompañadas de un deterioro intelectual grave.
En general, el pronóstico depende mucho del momento del inicio del tratamiento; cuando éste se inicia en fases tempranas el pronóstico es más favorable.
La epilepsia puede causar discapacidad de por vida. Si una persona desarrolla un trastorno de salud persistente como resultado de la enfermedad, que limita sus actividades cotidianas, esto se determina mediante un examen médico y social. Este también decide la asignación a un grupo de discapacidad específico. Primero debe contactar a su médico de cabecera sobre este tema, quien lo presentará a la comisión.