
Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
Síndrome de Treacher Collins
Médico experto del artículo.

Último revisado: 04.07.2025

Las alteraciones intrauterinas en los procesos de desarrollo óseo producen graves deformidades craneofaciales, y una de las variedades de dicha patología es el síndrome de Treacher Collins (STC) o disostosis mandibulofascial, es decir, maxilofacial.
Código de enfermedad según CIE 10: clase XVII (anomalías congénitas, deformaciones y trastornos cromosómicos), Q75.4 - disostosis mandibulofacial.
Causas Síndrome de Treacher Collins
Este síndrome debe su nombre al destacado oftalmólogo británico Edward Treacher Collins, quien describió las principales características de la patología hace más de cien años. Sin embargo, los médicos europeos suelen denominar a este tipo de anomalía ósea facial y mandibular enfermedad o síndrome de Franceschetti, basándose en la extensa investigación del oftalmólogo suizo Adolf Franceschetti, quien introdujo el término «disostosis mandibulofascial» a mediados del siglo pasado. En el ámbito médico, también se utiliza el nombre de síndrome de Franceschetti-Collins.
El síndrome de Treacher Collins está causado por mutaciones en el gen TCOF1 (en el locus cromosómico 5q31.3-33.3), que codifica una fosfoproteína nucleolar responsable de la formación de la parte craneofacial del embrión humano. Como resultado de una disminución prematura de la cantidad de esta proteína, se altera la biogénesis y las funciones del ARNr. Según genetistas del programa de investigación del Genoma Humano, estos procesos conducen a una reducción en la proliferación de células embrionarias de la cresta neural, una cresta a lo largo del surco neural que se cierra formando un tubo neural durante el desarrollo embrionario.
La formación de los tejidos faciales se produce debido a la transformación y diferenciación de las células de la parte superior (cabeza) de la cresta neural, que migran a lo largo del tubo neural hasta la zona del primer y segundo arco branquial del embrión. La deficiencia de estas células provoca deformaciones craneofaciales. El período crítico para la aparición de anomalías es de 18 a 28 días después de la fecundación. Tras completar la migración de las células de la cresta neural (en la cuarta semana de gestación), se forman casi todos los tejidos mesenquimales laxos en el área facial, que posteriormente (de 5 a 8 semanas) se diferencian en tejidos esqueléticos y conectivos de todas las partes de la cara, el cuello, la laringe, el oído (incluido el oído interno) y los futuros dientes.
Patogenesia
La patogenia del síndrome de Treacher Collins suele ser familiar, y la anomalía se hereda de forma autosómica dominante, aunque existen casos de transmisión autosómica recesiva del defecto (con mutaciones en otros genes, en particular, POLR1C y POLR1D). Lo más impredecible de la disostosis maxilofacial es que la mutación se hereda a los hijos solo en el 40-48% de los casos. Es decir, en el 52-60% de los pacientes, las causas del síndrome de Treacher Collins no se asocian con la presencia de una anomalía en la familia, y se cree que la patología se produce como resultado de mutaciones genéticas esporádicas de novo. Lo más probable es que las nuevas mutaciones sean consecuencia de efectos teratogénicos en el feto durante el embarazo.
Entre las causas teratogénicas de este síndrome, los expertos mencionan altas dosis de etanol (alcohol etílico), radiación, humo de cigarrillo, citomegavirus y toxoplasmosis, así como herbicidas a base de glifosato (Roundal, Glyfor, Tornado, etc.). La lista de factores iatrogénicos incluye medicamentos para el acné y la seborrea con ácido 13-cis-retinoico (isotretinoína, Accutane); el anticonvulsivo fenitoína (Dilantin, Epanutin); y los psicofármacos diazepam, valium, relanium y seduxen.
Síntomas Síndrome de Treacher Collins
En general, los signos clínicos de la disostosis mandibulofascial y su grado de expresión dependen de las características de las mutaciones genéticas. Los primeros signos de esta anomalía, en la mayoría de los casos, son visibles en el niño inmediatamente después del nacimiento: la cara con síndrome de Treacher-Collins presenta una apariencia característica. Además, las anomalías morfológicas suelen ser bilaterales y simétricas.
Los síntomas más evidentes del síndrome de Treacher Collins son:
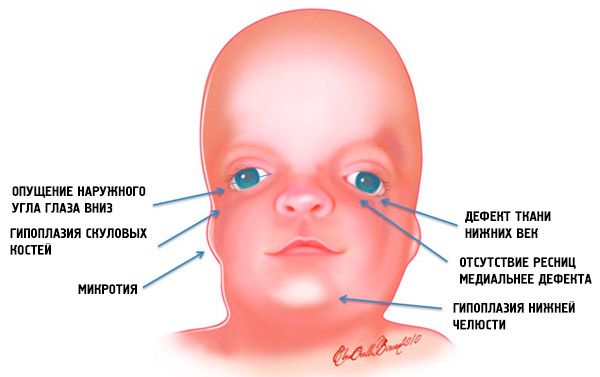
- subdesarrollo (hipoplasia) de los huesos faciales del cráneo: cigomático, procesos cigomáticos del hueso frontal, placas pterigoideas laterales, senos paranasales, mandíbula inferior y protuberancias de las epífisis óseas (cóndilos);
- subdesarrollo de los huesos de la mandíbula inferior (micrognatia) y un ángulo mandibular más obtuso de lo habitual;
- La nariz es de tamaño normal, pero parece grande debido a la hipoplasia de los arcos superciliares y al subdesarrollo o ausencia de los arcos cigomáticos en la región temporal;
- las hendiduras de los ojos están hacia abajo, es decir, la forma de los ojos es anormal, con las esquinas externas caídas hacia abajo;
- defectos de los párpados inferiores (coloboma) y ausencia parcial de pestañas en ellos;
- aurículas de forma irregular con una amplia gama de desviaciones, incluida su ubicación en la comisura de la mandíbula inferior, ausencia de lóbulos, fístulas ciegas entre el trago de la oreja y la comisura de la boca, etc.;
- estrechamiento o cierre (atresia) del canal auditivo externo y anomalías de los huesecillos del oído medio;
- ausencia o hipoplasia de las glándulas salivales parótidas;
- hipoplasia faríngea (estrechamiento de la faringe y las vías respiratorias);
- no fusión del paladar duro (paladar hendido), así como ausencia, acortamiento o inmovilidad del paladar blando.
Estas anomalías anatómicas presentan complicaciones en todos los casos. Se trata de trastornos auditivos funcionales, como pérdida auditiva conductiva o sordera total; discapacidad visual debido a la formación inadecuada de los globos oculares; defectos del paladar que causan dificultades para alimentarse y tragar. Existen trastornos de oclusión dental (maloclusión) asociados a defectos mandibulares, que a su vez causan problemas de masticación y articulación. Las patologías del paladar blando explican la voz nasal.
Complicaciones y consecuencias
Las consecuencias de las anomalías maxilofaciales en el síndrome de Treacher Collins son que al nacer las capacidades intelectuales del niño son normales, pero debido a los defectos auditivos y otros trastornos, se observa retraso mental secundario.
Además, los niños con tales defectos sienten profundamente su inferioridad y sufren, lo que afecta negativamente a su sistema nervioso y a su psique.
Diagnostico Síndrome de Treacher Collins
El diagnóstico posnatal del síndrome de Treacher Collins se basa fundamentalmente en los signos clínicos. La disostosis craneofacial se identifica fácilmente cuando el síndrome se manifiesta plenamente, pero cuando los síntomas patológicos son mínimos, pueden surgir dificultades para establecer un diagnóstico correcto.
En este caso, se debe prestar especial atención a la evaluación de todas las funciones asociadas con anomalías, especialmente las que afectan la respiración (debido al riesgo de apnea del sueño). También se debe evaluar y monitorizar la eficacia de la alimentación y la saturación de oxígeno de la hemoglobina.
Posteriormente, entre el quinto y sexto día después del nacimiento, será necesario determinar el grado de daño auditivo mediante pruebas audiológicas, que deberán realizarse en el hospital de maternidad.
Se prescribe un examen, durante el cual se realizan diagnósticos instrumentales mediante fluoroscopia de dismorfología craneofacial; pantomografía (radiografía panorámica de las estructuras óseas del cráneo facial); tomografía computarizada craneal completa en varias proyecciones; tomografía computarizada o resonancia magnética del cerebro para determinar el estado del canal auditivo interno.
El diagnóstico más temprano – prenatal – de anomalías maxilofaciales en presencia del síndrome de Treacher Collins en la historia familiar es posible mediante una biopsia de vellosidades coriónicas a las 10-11 semanas de embarazo (el procedimiento amenaza con aborto e infección del útero).
También se toman análisis de sangre a los familiares; a las 16-17 semanas de embarazo se analiza el líquido amniótico (amniocentesis transabdominal); a las 18-20 semanas de embarazo se realiza una fetoscopia y se extrae sangre de los vasos fetales de la placenta.
Pero lo más frecuente es que la ecografía se utilice en el diagnóstico prenatal de este síndrome en el feto (a las 20-24 semanas de embarazo).
¿Qué pruebas son necesarias?
Diagnóstico diferencial
Estos mismos métodos son utilizados por los especialistas cuando se necesitan diagnósticos diferenciales para reconocer el síndrome leve de Treacher Collins y distinguirlo de otras anomalías congénitas de los huesos craneofaciales, en particular: síndromes de Apert, Crouzon, Nager, Peters-Hewels, Hellermann-Steph, así como microsomía hemifacial (síndrome de Goldenhar), hipertelorismo, fusión prematura de las suturas craneales (craneosinostosis) o fusión alterada de los huesos faciales (craneosinostosis).
Tratamiento Síndrome de Treacher Collins
Como en todos los casos de defectos congénitos de origen genético, el tratamiento de las formas graves del síndrome de Treacher-Collins es exclusivamente paliativo, ya que no existen métodos terapéuticos para estas patologías. El espectro y el grado de malformaciones en este síndrome son amplios y, por lo tanto, la naturaleza e intensidad de la intervención médica también ofrecen diversas opciones.
Los audífonos se utilizan para corregir y mejorar la audición, y las sesiones de terapia del habla se utilizan para mejorar el habla.
En casos graves de estrechamiento de las vías respiratorias (traqueotomía) y de laringe (gastrostomía para la alimentación), se requieren intervenciones quirúrgicas a una edad temprana. También puede ser necesaria la corrección quirúrgica del paladar.
Las cirugías de alargamiento mandibular se realizan a los 2-3 años o más. La reconstrucción de tejidos blandos incluye la corrección del coloboma del párpado inferior y la cirugía plástica auricular.
Prevención
La prevención del síndrome de Treacher Collins implica que los futuros padres acudan a asesoramiento genético y, si existen antecedentes familiares del síndrome, surge la cuestión de la posibilidad del propio embarazo, para evitar el nacimiento de un niño con anomalías craneofaciales.
Pronóstico
¿Cuál es el pronóstico de esta patología? Depende del grado de deformación y de la intensidad de los síntomas. El síndrome de Treacher Collins es un diagnóstico de por vida.
 [ 25 ]
[ 25 ]