
Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
Síndrome de Angelman en niños y adultos
Médico experto del artículo.

Último revisado: 04.07.2025

Hay varias enfermedades para las que expresiones como "cuídate y no te enfermarás" suenan, como mínimo, ridículas. Se trata de patologías en las que algunas anomalías mentales y físicas son inherentes al cuerpo del niño incluso antes del nacimiento, pero los padres no tienen la culpa. Estas enfermedades son causadas por mutaciones o anomalías en los conjuntos de cromosomas y se denominan cromosómicas o genéticas. El síndrome de Angelman, el síndrome de Down, el síndrome de Patau, el síndrome de Edwards, el síndrome de Turner y el síndrome de Prader-Willi son solo una parte de una lista bastante amplia de enfermedades genéticas.
Síndrome del hombre feliz
En esta ocasión, hablaremos de la patología que lleva el nombre del pediatra inglés Harry Angelman, quien planteó por primera vez este problema en 1965, tras haber encontrado en su consulta el día anterior a tres niños inusuales, unidos por síntomas peculiares comunes. El médico los llamó "niños-muñecos" y escribió un artículo sobre ellos, inicialmente titulado "Niños-marionetas". El artículo y su título se inspiraron en una pintura expuesta en un museo de Verona. La pintura representaba a un niño riendo y se titulaba "El niño títere". La asociación del niño representado en la pintura con los tres niños que Angelman atendió en su consulta impulsó al pediatra a agruparlos en un solo grupo debido a la enfermedad que padecían.
No es sorprendente que los niños mencionados en el artículo no fueran detectados por otros médicos. Al fin y al cabo, a primera vista parecía que padecían enfermedades completamente distintas, tan distinto era el cuadro clínico general en tres casos distintos. Quizás la "nueva" patología cromosómica habría interesado a otros científicos, pero en aquel entonces la genética aún no estaba lo suficientemente desarrollada como para confirmar la hipótesis del médico inglés. Por lo tanto, tras cierto interés, el artículo quedó relegado a un segundo plano durante mucho tiempo.
La siguiente mención del síndrome de Angelman, como se denominó ahora el artículo del pediatra inglés G. Angelman, data de principios de los años 80 del siglo XX. Y solo en 1987 se descubrió la razón por la que un pequeño porcentaje de niños nace con tales anomalías que, desde fuera, parecen estar constantemente sonriendo y felices. De hecho, esto no es cierto en absoluto, y la sonrisa es solo una mueca tras la cual se esconden la tristeza y el dolor de los padres.
Epidemiología
Según las estadísticas, una mutación cromosómica en un niño puede desarrollarse tanto en el contexto de mutaciones similares en sus padres como en ausencia de ellas. No existe una naturaleza hereditaria clara del síndrome de Angelman (SA), pero la probabilidad de desarrollar la patología en padres con mutaciones cromosómicas es bastante alta.
También es interesante que si una familia ya tiene un hijo con SA, existe un uno por ciento de posibilidades de tener un segundo hijo con el mismo trastorno, incluso si los padres están sanos.
Aún no existen estadísticas exactas sobre el número de pacientes con síndrome de Angelman. Quizás la razón sea la variedad de síntomas, que pueden presentarse con cierta composición o no presentarse durante mucho tiempo. Se estima que la prevalencia de la enfermedad es de 1 niño por cada 20.000 recién nacidos. Sin embargo, esta cifra es muy aproximada.
Causas Síndrome de Angelman
El síndrome de Angelman es el nombre médico de una patología cromosómica, pero no es la única. Se le conoce como síndrome del niño muñeca, síndrome de la marioneta feliz, síndrome de Petrushka y síndrome de la muñeca riendo. Se inventan todo tipo de nombres (a veces incluso ofensivos para los propios pacientes y sus padres), pero una enfermedad es una enfermedad, por muy graciosa que parezca y sin importar las razones.
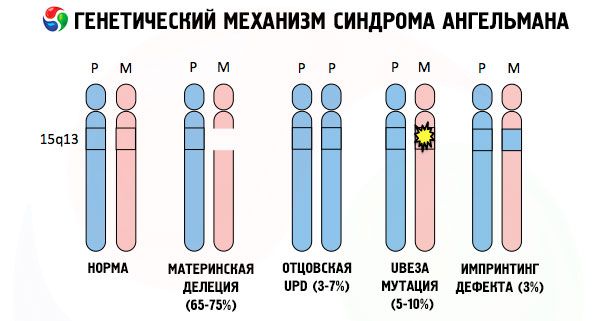
Las causas del desarrollo del síndrome de Angelman, como muchas otras patologías genéticas, son en todos los casos alteraciones en la estructura de uno de los cromosomas o del conjunto cromosómico en su conjunto. Pero en nuestro caso, el problema radica en el cromosoma 15, transmitido por la madre. Es decir, el cromosoma paterno en este caso no presenta anomalías, pero el femenino sufre ciertas mutaciones.
Según el tipo de anomalía cromosómica, el síndrome de Angelman se clasifica como una mutación cromosómica. Estas mutaciones se consideran:
- Una deleción (ausencia de una sección de un cromosoma que contiene un determinado conjunto de genes; si falta uno de los genes, hablamos de una microdeleción), que es el resultado de dos roturas y una reunión, cuando se pierde una sección del cromosoma original.
- Duplicación (presencia de una sección extra en un cromosoma que es una copia de una existente), que en la mayoría de los casos conduce a la muerte de una persona y, con menor frecuencia, a la infertilidad.
- Inversión (inversión de una de las secciones del cromosoma en 180 grados, es decir, en la dirección opuesta, y entonces los genes en ella se ubican en el orden opuesto), cuando los extremos rotos del cromosoma se conectan en un orden diferente al original.
- Inserción (si parte del material genético de un cromosoma está fuera de lugar),
- translocación (si una determinada sección de un cromosoma está unida a otro cromosoma; dicha mutación puede ser mutua sin pérdida de secciones).
Al recibir un cromosoma mutado de una madre desprevenida, el niño está condenado a nacer con anomalías. La causa más común del síndrome de Angelman aún se considera una deleción del cromosoma 15 materno, cuando falta una pequeña sección. Se consideran mutaciones menos comunes en el síndrome de la "muñeca sonriente":
- translocación,
- disomía unipaternal (si el niño recibió un par de cromosomas del padre, el cromosoma materno está ausente),
- mutación de genes en el ADN, que son a la vez el principal material de construcción (genético) y las instrucciones para su correcto uso (en particular, la mutación del gen ube3a en el cromosoma materno).
La presencia de una de estas mutaciones en los padres es un factor de riesgo para el desarrollo del síndrome de Angelman en los hijos. Sin embargo, no solo las mutaciones cromosómicas, sino también las genómicas (que se asocian con un cambio cuantitativo en el conjunto de cromosomas y son más comunes que las cromosómicas) pueden provocar el desarrollo de la enfermedad en un niño. Una mutación genómica común incluye la trisomía cromosómica (si el conjunto de cromosomas de una persona tiene más de 46 cromosomas).
Para que una patología se presente en un niño, no es necesario que los padres presenten anomalías cromosómicas. Sin embargo, existe un cierto porcentaje de pacientes cuya enfermedad es hereditaria.
Patogenesia
Profundicemos un poco más en la biología, o más precisamente, en la genética. La información genética de cada organismo humano se encuentra en 23 pares de cromosomas. Un cromosoma de cada par se transmite al hijo del padre, y el otro, de la madre. Todos los pares de cromosomas difieren en forma y tamaño y contienen cierta información. Así, el par 23 de cromosomas (X e Y) es responsable de la formación de las características sexuales del bebé (XX: niña, XY: niño, mientras que el cromosoma Y solo puede ser heredado por el hijo del padre).
Idealmente, un niño recibe 46 cromosomas de sus padres, los cuales conforman sus características genéticas y lo predeterminan como individuo. Un número mayor de cromosomas se denomina trisomía y se considera una desviación de la norma. Por ejemplo, la presencia del cromosoma 47 en el conjunto cromosómico (cariotipo, que determina la especie y las características individuales) causa la aparición del síndrome de Down.
Si los cromosomas se tiñen con un tinte especial, al microscopio se pueden observar franjas de diferentes tonos a lo largo de cada uno de ellos. Dentro de cada franja se encuentra una gran cantidad de genes. Todas estas franjas están numeradas por los científicos y tienen una ubicación fija. La ausencia de una franja se considera una desviación de la norma. En el síndrome de Angelman, es frecuente observar la ausencia de segmentos del cromosoma materno en el intervalo q11-q13, ubicado en el brazo largo, cuyo número de bases de ADN es de tan solo unos 4 millones.
Se considera que el componente principal del cromosoma es una molécula de ADN increíblemente larga que contiene miles de genes y decenas y cientos de millones de bases nitrogenadas. Así, el cromosoma 15, responsable del desarrollo del síndrome de Angelman y varios otros, contiene 1200 genes y alrededor de 100 millones de bases. Cualquier alteración en la estructura de la molécula de ADN afectará sin duda la apariencia y el desarrollo del futuro niño.
La información genética contenida en los genes se convierte en proteína o ARN. Este proceso se denomina expresión génica. De esta manera, la información genética recibida de los padres adquiere forma y contenido, que se materializa en su heredero único, ya sea hombre o mujer.
Existen diversas patologías con un tipo de herencia no clásico, entre ellas el síndrome de Angelman, en el que los genes recibidos de los padres como parte de cromosomas pareados llevan una impronta única de los padres y se manifiestan de formas diferentes.
Así pues, el síndrome de Angelman es un ejemplo notable de impronta genómica, en el que la expresión génica en el cuerpo del niño depende directamente del progenitor del que provienen los alelos (diferentes formas de un mismo gen, recibidas del padre y la madre, ubicadas en secciones idénticas de cromosomas pareados). Es decir, solo las anomalías en el cromosoma materno conducen al desarrollo del síndrome, mientras que las mutaciones y los trastornos estructurales del cromosoma paterno causan patologías completamente diferentes.
En esta patología, existe una carencia de ciertos genes en el cromosoma materno o una pérdida o reducción de la actividad de genes individuales (en la gran mayoría de los casos, el gen ube3a, que participa en el metabolismo de la ubiquitina, una proteína que regula la degradación de otras proteínas). Como resultado, se diagnostica al niño con anomalías del desarrollo mental y deformidades físicas.
Síntomas Síndrome de Angelman
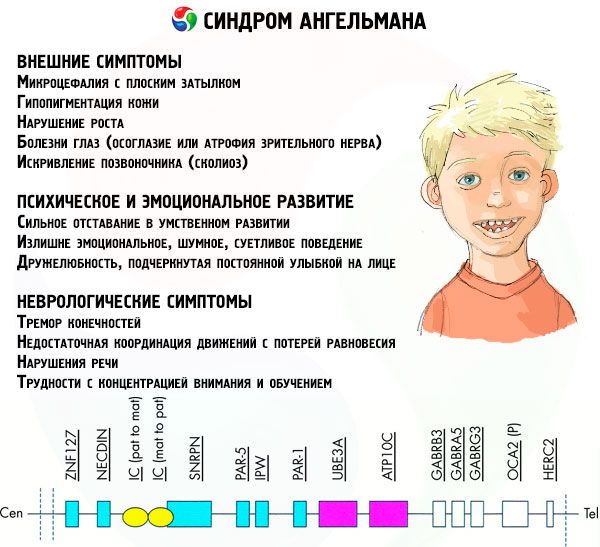
Los síntomas del síndrome de Angelman afectan diversos aspectos de la vida y el desarrollo del niño: físico, neurológico y mental. En base a esto, se pueden identificar tres grupos de síntomas que indican el desarrollo de esta patología.
- Síntomas externos o físicos:
- una cabeza desproporcionadamente pequeña en comparación con el cuerpo y las extremidades, que son de tamaño normal,
- boca demasiado ancha,
- Casi siempre hay una sonrisa en la cara (con la boca abierta),
- dientes escasos,
- labio superior estrecho,
- lengua ancha que sobresale con frecuencia,
- mandíbula inferior prominente,
- barbilla puntiaguda,
- piel muy clara, a menudo con pelo (albinismo, asociado al hecho de que el cuerpo no produce el pigmento melanina),
- Manchas oscuras en piel clara (hipopigmentación debido a una producción insuficiente de melanina)
- síntomas físicos o externos: enfermedades oculares como estrabismo o atrofia del nervio óptico,
- curvatura de la columna vertebral (escoliosis),
- piernas rígidas (al caminar, una persona no dobla las piernas a la altura de las rodillas debido a la baja movilidad de las articulaciones, de ahí la comparación con el andar de una muñeca).
- Síntomas relacionados con el desarrollo mental y emocional:
- retraso mental grave,
- comportamiento excesivamente emocional, ruidoso y quisquilloso,
- aplausos frecuentes,
- expresó amabilidad, enfatizada por una sonrisa constante en el rostro,
- Risas frecuentes sin motivo.
- Síntomas neurológicos:
- temblor de las extremidades,
- coordinación insuficiente de movimientos con pérdida del equilibrio,
- disminución del tono muscular,
- diversos trastornos del sueño,
- frecuentes ataques histéricos en la infancia,
- Trastornos del habla (el niño empieza a hablar tarde, tiene malas habilidades de comunicación y habla arrastrada),
- hiperactividad en el contexto de una mayor excitabilidad,
- dificultades para concentrarse y aprender.
Pero este es un panorama general de la enfermedad. De hecho, el cuadro clínico del síndrome de Angelman depende en gran medida de la etapa de desarrollo de la enfermedad y del tipo de mutación cromosómica que la causó. Esto significa que los síntomas de la enfermedad pueden variar significativamente entre pacientes, lo que durante mucho tiempo impidió distinguir esta patología de otras con un cuadro clínico similar.
Entre el total de síntomas podemos destacar aquellos que son característicos de todos los pacientes sin excepción:
- retraso mental grave,
- comportamiento inadecuado (risa irrazonable, aumento de la excitabilidad, falta de concentración, estado de euforia),
- subdesarrollo de las habilidades motoras,
- Mala coordinación de movimientos, ataxia de la marcha (ritmo irregular, balanceo de lado a lado, etc.), temblor de las extremidades.
- Trastorno del desarrollo del habla con predominio de medios de comunicación no verbales.
Entre los síntomas que presentan la gran mayoría de los pacientes se pueden distinguir los siguientes:
- desproporción entre la cabeza y el cuerpo causada por un retraso en el desarrollo físico,
- En muchos pacientes la forma del cráneo es tal que el tamaño del cerebro permanece menor que en personas sanas (microcefalia),
- convulsiones epilépticas antes de los 3 años de edad con una disminución progresiva de la fuerza y la frecuencia a mayor edad,
- distorsión de los parámetros EEG (fluctuaciones y alta amplitud de ondas de baja frecuencia).
Estos síntomas son bastante comunes, sin embargo, el 20% de los pacientes con síndrome de Angelman no los presentan.
Con menor frecuencia aún se pueden diagnosticar manifestaciones de la enfermedad como:
- estrabismo severo o leve,
- control deficiente del movimiento de la lengua, lo que hace que los pacientes a menudo saquen la lengua sin motivo alguno,
- Dificultades para tragar y succionar, especialmente en niños pequeños.
- alteración de la pigmentación de la piel y los ojos,
- brazos levantados o doblados al caminar,
- hiperreflexia,
- Trastornos del sueño, especialmente en la infancia,
- salivación frecuente,
- sed insaciable,
- movimientos de masticación excesivamente activos,
- hipersensibilidad al calor,
- parte posterior plana de la cabeza,
- mandíbula inferior prominente,
- palmas lisas.
Un gran porcentaje de pacientes presenta problemas para orinar, que controlan mal, deterioro de la motricidad fina, lo que dificulta el autocuidado y el aprendizaje, y sobrepeso. Casi todos los pacientes llegan a la pubertad más tarde que sus pares sanos.
Los niños con síndrome de Angelman perciben bien el habla oral y la comprenden, pero no desean participar en conversaciones, limitándose a unas pocas decenas de palabras necesarias en la vida cotidiana. Sin embargo, en la edad adulta, estos pacientes parecen más jóvenes que sus compañeros sin patologías genéticas.
Muchos síntomas del síndrome de Angelman son inconstantes, por lo que el cuadro clínico de la enfermedad cambia significativamente con la edad. Las convulsiones y los ataques epilépticos se vuelven menos frecuentes o desaparecen por completo, el paciente se vuelve menos excitable y el sueño mejora.
Complicaciones y consecuencias
El síndrome de Angelman es una cromosómica grave, actualmente prácticamente incurable, que priva a los pacientes de la oportunidad de llevar una vida normal. La vida de un niño con SA depende en gran medida del tipo de anomalía cromosómica.
La duplicación de un segmento cromosómico es incompatible con la vida en la mayoría de los casos. Incluso si estos pacientes no mueren en la infancia y llegan a la pubertad, no tienen posibilidad de tener hijos.
La deleción o ausencia de una parte de los genes, que se presenta con mayor frecuencia en el síndrome de Angelman, dificulta que el niño aprenda a caminar y hablar. Estos niños presentan una forma más grave de retraso mental, y las convulsiones epilépticas son más frecuentes y de mayor intensidad que en pacientes con otras anomalías cromosómicas.
Si solo hay una mutación de un gen, con la debida atención y enfoque se le pueden enseñar al niño los conceptos básicos del autocuidado, la comunicación y la interacción en un grupo, aunque todavía estará por detrás de sus compañeros en el desarrollo.
Para los niños con síndrome de Angelman, que son amables por naturaleza, lo más importante es el amor y la atención de sus padres. Solo así su educación dará frutos, incluso si es pequeña. Por supuesto, los pacientes con SA no podrán asistir a una escuela regular. Necesitan clases especiales donde primero se les enseñe a concentrarse y luego, gradualmente, se les impartan los conocimientos básicos de la escuela.
Diagnostico Síndrome de Angelman
El síndrome de Angelman es una patología congénita del desarrollo. Sin embargo, debido a ciertas circunstancias, a menudo es imposible diagnosticarlo en la infancia y la primera infancia. Esto se debe a la inespecificidad y la escasa expresión de los síntomas en bebés y niños menores de 3 años. Además, la prevalencia de la enfermedad en nuestro país no es tan alta como para que los médicos hayan aprendido a reconocerla entre sus pares.
El síndrome de Angelman en bebés puede manifestarse como una disminución del tono muscular, lo que se traduce en problemas de alimentación (debilidad del reflejo de succión y deglución) y, posteriormente, dificultades para aprender a caminar (estos niños empiezan a caminar mucho más tarde). Estos síntomas son los primeros signos de una anomalía del desarrollo en el bebé, que bien podría estar asociada a una anomalía cromosómica. Solo el análisis genético puede confirmar esta hipótesis.
Se presta especial atención a los niños cuyos padres presentan diversos trastornos genómicos o cromosómicos. Al fin y al cabo, la enfermedad puede no manifestarse inicialmente, y si se detecta a tiempo, al comenzar a trabajar intensamente con el niño, es posible lograr un éxito académico significativamente mayor, ralentizando así su progresión.
Si los padres presentan diversas anomalías cromosómicas, el análisis genético se realiza incluso antes de que nazca el bebé, ya que la SA es una de las patologías que se pueden detectar en la etapa embrionaria.
La recolección de material para investigación genética se puede realizar de dos maneras:
- invasiva (con un cierto porcentaje de riesgo, ya que es necesario penetrar el útero para tomar una muestra de líquido amniótico),
- no invasivo (análisis del ADN del bebé a partir de la sangre de la madre).
A continuación se realizan los siguientes estudios:
- hibridación in situ fluorescente (método FISH): unión de una sonda de ADN marcada con un colorante especial al ADN en estudio, seguida de su examen bajo un microscopio.
- análisis de mutaciones en el gen ube3a y genes impresos,
- Análisis de la metilación del ADN mediante métodos especiales utilizados en genética.
Las pruebas genéticas proporcionan información bastante precisa en caso de anomalías cromosómicas, lo que significa que los futuros padres saben de antemano qué esperar. Sin embargo, hay excepciones. En cierto grupo de pacientes, ante la presencia de todos los síntomas indicativos de patología, los resultados de la prueba se mantienen normales. Es decir, la patología solo puede identificarse mediante una observación cuidadosa del niño desde la primera infancia: cómo come, cuándo empezó a caminar y hablar, si dobla las piernas al caminar, etc.
Además del método FISH, entre los métodos de diagnóstico instrumental del síndrome de Angelman se pueden destacar la tomografía (TC o RMN), que ayuda a determinar el estado y el tamaño del cerebro, y el electroencefalograma (EEG), que muestra cómo funcionan las distintas partes del cerebro.
Los médicos suelen hacer el diagnóstico definitivo a la edad de 3-7 años, cuando el paciente ya presenta la mayoría de los síntomas y es visible la dinámica del desarrollo de la enfermedad.
¿Qué pruebas son necesarias?
Diagnóstico diferencial
El síndrome de Angelman es una patología genética que prácticamente no presenta manifestaciones específicas. La mayoría de los síntomas pueden indicar tanto el SA como otras patologías genéticas.
El diagnóstico diferencial del síndrome de Angelman se realiza con las siguientes patologías:
- Síndrome de Pitt-Hopkins (los pacientes se caracterizan por retraso mental, carácter alegre, sonrisas, boca grande y ancha, y se observa microcefalia). La diferencia radica en episodios de hiperventilación y apnea en estado de vigilia.
- Síndrome de Christianson (los pacientes son personas con retraso mental, de carácter alegre, incapaces de hablar, caracterizados por microcefalia, ataxia, convulsiones, movimientos musculares involuntarios).
- Síndrome de Mowat-Wilson (síntomas: retraso mental, convulsiones epilépticas, mentón prominente, boca abierta, expresión facial alegre, microcefalia). Características: gran distancia entre los ojos, ojos oblicuos hacia adentro, punta de la nariz redondeada, pabellón auricular invertido.
- Síndrome de Kabuki (caracterizado por retraso mental leve a moderado, problemas del habla y motricidad, debilidad muscular, convulsiones epilépticas, microcefalia, intervalos largos entre picores y alteración de la coordinación). Se caracteriza por cejas arqueadas, porción lateral del párpado inferior evertida, ojos muy separados, fisuras palpebrales largas con pestañas largas y espesas.
- Síndrome de Rett (diferenciación del síndrome de Rett en mujeres). Síntomas: retraso en el desarrollo del habla, convulsiones, microcefalia. La diferencia radica en la ausencia de expresión facial alegre y en los episodios de apnea y apraxia que progresan con el tiempo.
- Síndrome de retraso mental autosómico recesivo 38 (síntomas: retraso mental marcado con retraso en las habilidades motoras y el habla, debilidad muscular, problemas de alimentación en la infancia, impulsividad). El rasgo distintivo es el color azul del iris.
- Síndrome de duplicación del gen MECP2 (diferenciación del SA en varones). Síntomas: retraso mental grave, debilidad muscular desde la infancia, problemas o ausencia del habla, epilepsia. Características distintivas: miopatía progresiva, infecciones recurrentes.
- Síndrome de Kleefstra (síntomas: problemas del habla y el pensamiento, debilidad muscular, alteraciones del sueño, falta de atención, boca abierta, hiperactividad, convulsiones, ataxia, trastornos del equilibrio). Rasgos distintivos: cara plana, nariz corta y respingada, ojos separados, labio inferior grande y evertido, arrebatos de agresividad.
- Síndrome de Smith-Magenis (caracterizado por convulsiones, problemas de sueño y trastornos del desarrollo intelectual y motor). Entre sus rasgos distintivos se incluyen una cara ancha y plana, y una frente prominente.
- Síndrome de Koolen-de Vries (retardo mental leve a moderado, debilidad muscular, convulsiones, amabilidad). Rasgos distintivos: cara alargada con frente alta, orejas prominentes, ojos rasgados, alta movilidad articular, cardiopatías congénitas.
- Síndrome de Phelan-McDermid (síntomas: retraso mental, trastornos del habla o ausencia del habla). Características: manos grandes con músculos desarrollados, debilidad muscular de nacimiento, sudoración débil.
Patologías como la deficiencia de adenilsuccinato, el síndrome de retraso mental autosómico recesivo tipo 1, el síndrome de duplicación del cromosoma 2q23.1, los síndromes de haploinsuficiencia de los genes FOXG1, STXBP1 o MEF2C y algunas otras pueden “presumir” de síntomas similares al síndrome de Angelman.
La tarea del médico es hacer un diagnóstico preciso, diferenciar el síndrome de Angelman de patologías con síntomas similares y prescribir un tratamiento eficaz que sea relevante para la etapa diagnosticada de la enfermedad.
Tratamiento Síndrome de Angelman
El síndrome de Angelman es una de esas patologías para las que la medicina aún busca un tratamiento eficaz. El tratamiento etiológico de la enfermedad se encuentra en fase de desarrollo con diversos métodos y medios, muchos de los cuales aún no se han probado en humanos. Esto significa que, por ahora, los médicos deben limitarse a la terapia sintomática, que ayuda a aliviar la difícil situación de niños y adultos con síndrome de marioneta, que sufren convulsiones epilépticas, salivación, hipotensión y trastornos del sueño.
Por lo tanto, es posible reducir la frecuencia e intensidad de las crisis epilépticas con un anticonvulsivo adecuado. Sin embargo, la dificultad radica en que las crisis en pacientes con SA difieren de las crisis epilépticas comunes en que se caracterizan por varios tipos de convulsiones, lo que significa que la afección puede aliviarse administrando varios fármacos simultáneamente.
Los anticonvulsivos más populares para tratar el síndrome de Angelman son: ácido valproico, topiramato, lamotrigina, levetiracetam, clonazepam y fármacos derivados. Con menos frecuencia se utilizan fármacos derivados de carmazepina, fenitoína, fenobarbital y etosuximida, ya que algunos de ellos pueden provocar un efecto paradójico que consiste en intensificar y aumentar la frecuencia de las crisis epilépticas. Esto ocurre si el fármaco se utiliza como parte de la monoterapia.
Para tratar el babeo, se suelen utilizar dos métodos: medicamentos (fármacos que suprimen la producción de saliva) y cirugía, que consiste en la reimplantación de los conductos salivales. Sin embargo, en el caso de la salivación espontánea, estos métodos se consideran ineficaces y el tema sigue abierto. Los padres y quienes cuidan de estos pacientes deben prestar especial atención a este problema, ya que los pacientes no suelen controlar el babeo y algunos simplemente son incapaces de cuidar de sí mismos.
Otro problema es la corta duración del sueño. A menudo, los niños con síndrome de Angelman no duermen más de 5 horas, lo que afecta negativamente el funcionamiento de todo el cuerpo. Los niños fácilmente excitables y activos, amantes de los juegos y la comunicación (aunque intenten limitarse a métodos no verbales), se sienten notablemente cansados durante el día. Para descansar bien, el cuerpo necesita un sueño profundo y completo, pero precisamente ahí reside el problema.
Parecería que los sedantes (fenotiazinas y antipsicóticos atípicos) que calman el sistema nervioso deberían ser suficientes para mejorar el sueño en pacientes excitables. Sin embargo, en el caso de la EA, el uso de estos fármacos conlleva la aparición de efectos negativos. Por lo tanto, los médicos siguen prefiriendo somníferos suaves, como la melatonina (un fármaco hormonal natural basado en la hormona del sueño), que se administra a los pacientes una hora antes de acostarse en una dosis de un comprimido, y la difenhidramina. La frecuencia de administración y la dosis las determina el médico en función de la afección y la edad del paciente.
A veces, los pacientes con síndrome de Angelman tienen problemas digestivos y de evacuación. Puede mejorar sus heces con laxantes (preferiblemente herbales).
O se puede abordar el problema de otra manera, como lo hicieron los médicos estadounidenses, basándose en algunos métodos de tratamiento del autismo, ya que muchos síntomas característicos del SA también lo son (impulsividad, movimientos involuntarios, acciones repetitivas, déficit de atención, problemas de comunicación, etc.). Se ha observado que la administración de la hormona secretina, que normaliza la digestión y las heces, tiene un efecto positivo en la atención de los pacientes, y la oxitocina ayuda a mejorar las capacidades cognitivas y la memoria del niño, así como a un comportamiento correcto.
Es cierto que las hormonas por sí solas no son suficientes, especialmente en el caso de los niños. En el síndrome de Angelman, se recomienda la terapia conductual, el trabajo con un psicólogo y un logopeda (enseñando métodos de comunicación no verbal y lenguaje de señas). La educación de estos niños debe basarse en un programa individualizado con la participación de maestros con formación específica, un psicólogo y los padres. Lamentablemente, esto no es posible en todas partes, y las familias se ven abandonadas a su propio problema.
Dado que muchos pacientes jóvenes con EA presentan bajo tono muscular y problemas articulares, se presta mucha atención a la fisioterapia. Con frecuencia, los médicos recurren a aplicaciones de parafina, electroforesis y magnetoterapia.
El masaje tónico activo y los ejercicios especiales de fisioterapia ayudarán al niño enfermo a ponerse de pie y caminar con seguridad después de un tiempo. La aquagymnasia es especialmente útil en este sentido, y se recomienda para la acuaerólisis en agua fría. Aumenta el tono muscular y enseña al niño a controlar su cuerpo y coordinar sus movimientos.
Tratamiento anticonvulsivo
El síntoma más peligroso del síndrome de Angelman son las convulsiones similares a las de la epilepsia. Este síntoma se observa en el 80 % de los pacientes, por lo que a todos ellos se les debe recetar un tratamiento anticonvulsivo eficaz.
El tratamiento de las convulsiones epilépticas se realiza con vitaminas y anticonvulsivos. En el síndrome de Angelman, acompañado de un síndrome convulsivo, serán útiles las vitaminas del grupo B, así como las vitaminas C, D y E. Sin embargo, automedicarse con vitaminas en este caso es muy peligroso, ya que la ingesta incontrolada de vitaminas puede reducir la eficacia de los antiepilépticos y provocar nuevas convulsiones más graves y prolongadas.
La selección de los anticonvulsivos y la prescripción de su dosis eficaz también deben ser realizadas por un médico especialista. Este médico también decide si un solo fármaco será suficiente o si el paciente deberá tomar dos o más fármacos durante un periodo prolongado.
Para la mayoría de los pacientes, los médicos prescriben medicamentos con ácido valproico (ácido valproico, Depakine, Convulex, Valparin, etc.), que previenen las convulsiones y mejoran el estado de ánimo y el estado mental de los pacientes.
El ácido valproico está disponible en comprimidos, jarabe y soluciones inyectables. El fármaco más popular es "Depakine", de liberación prolongada, en comprimidos y solución intravenosa. La dosis la determina el médico individualmente, dependiendo del peso, la edad y el estado del paciente.
El medicamento se toma con las comidas de 2 a 3 veces al día. La dosis diaria promedio es de 20 a 30 mg por kilogramo de peso corporal, con un máximo de 50 mg/kg al día.
Contraindicaciones de uso: No usar en caso de disfunción hepática y pancreática, diátesis hemorrágica, hepatitis, porfiria e hipersensibilidad al medicamento.
Los efectos secundarios incluyen temblores en las manos, trastornos digestivos y fecales y cambios en el peso corporal.
El topiramato también es un fármaco de elección para la apnea obstructiva del sueño. Se presenta en comprimidos y se utiliza tanto en monoterapia como en combinación con otros fármacos.
Método de administración y dosificación. Tome los comprimidos por vía oral, independientemente de la ingesta de alimentos. La dosis diaria inicial para adultos es de 25-50 mg, y para niños, de 0,5-1 mg/kg. La dosis se aumenta semanalmente según las indicaciones del médico.
Este medicamento no debe tomarse durante el embarazo ni la lactancia, ni en caso de hipersensibilidad a sus componentes. Presenta numerosos efectos secundarios.
Medicamentos que un médico puede recetar para el síndrome de Angelman: Clomazepam, Rivotril, Lamotrigina, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Medicina tradicional y homeopatía
La medicina tradicional, como las preparaciones homeopáticas, son por supuesto relativamente seguras, pero la efectividad de dicho tratamiento para el síndrome de Angelman puede considerarse controvertida.
Aunque el tratamiento tradicional puede ser útil en algunos casos, como por ejemplo, detener las convulsiones epilépticas, el tratamiento a base de hierbas puede ser bastante eficaz.
Una combinación medicinal a base de peonía, regaliz y lenteja de agua (los componentes se toman en cantidades iguales) produce buenos resultados. Las hierbas deben molerse hasta obtener harina. Tras dos semanas de tomarla, se puede notar una disminución significativa en la frecuencia de las convulsiones.
La decocción de lavanda (1 cucharadita por vaso de agua hirviendo) también es útil para los cólicos. La mezcla se hierve durante 5 minutos y se deja en infusión durante media hora. El medicamento se toma por la noche durante 14 días.
Una infusión acuosa (o alcohólica) de agripalma se considera eficaz para las convulsiones epilépticas.
Entre los preparados homeopáticos para prevenir las convulsiones en el síndrome de Angelman, se pueden utilizar medicamentos a base de manzanilla y agripalma, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum y Arsenicum album. Sin embargo, debe tenerse en cuenta que solo un médico homeópata puede recetar dosis eficaces y seguras de medicamentos en cada caso específico.
Prevención
Como el lector probablemente ya habrá comprendido, la medicina aún no puede prevenir las mutaciones genéticas ni otras anomalías cromosómicas, ni corregir la situación. Esto puede ocurrirle a cualquiera, ya que los niños con síndrome de Angelman nacen de padres sanos, y la genética, actualmente una de las ramas de la medicina menos estudiadas, aún no puede explicarlo.
Lo único que se puede hacer es planificar el embarazo de forma responsable, registrarse y someterse a las revisiones a tiempo. Sin embargo, como cualquier revisión, esta medida será más educativa que preventiva. Los padres jóvenes sabrán de antemano qué preparar y, en caso de una respuesta positiva, decidirán si pueden asumir la responsabilidad de criar a un hijo enfermo.
Pronóstico
El pronóstico del síndrome de Angelman depende de la naturaleza de la anomalía cromosómica y de la prontitud de su detección. Los más afectados son aquellos niños cuyo cromosoma 15 contiene lagunas genéticas (deleción). La probabilidad de que estos pacientes caminen y hablen es extremadamente baja. Otros casos pueden corregirse con un enfoque cuidadoso y cariño hacia su hijo.
Desafortunadamente, estos pacientes no podrán integrarse plenamente en la sociedad, a pesar de que no son nada tontos y comprenden el lenguaje y su significado. Sin embargo, tendrán problemas de comunicación el resto de sus vidas. Se les puede enseñar lenguaje de señas desde la infancia, pero no se les puede obligar a comunicarse con palabras. El vocabulario de los pacientes "hablantes" se limita al mínimo de palabras de uso cotidiano (5-15 palabras).
En cuanto a la esperanza de vida y la salud general de los pacientes con síndrome de Angelman, las cifras fluctúan en torno a valores promedio. En la edad adulta, los pacientes suelen presentar problemas de salud como escoliosis y obesidad, que, con un tratamiento adecuado, no ponen en peligro la vida.