
Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
Síndrome de Martin-Bell
Médico experto del artículo.

Último revisado: 04.07.2025
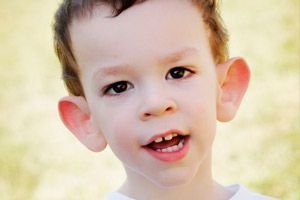
El síndrome de Martin-Bell fue descrito en 1943 por médicos, de quienes tomó su nombre. Esta enfermedad es un trastorno genético que consiste en retraso mental. En 1969, se identificaron cambios en el cromosoma X (fragilidad en el brazo distal) característicos de esta enfermedad. En 1991, los científicos descubrieron el gen responsable de su desarrollo. Esta enfermedad también se denomina "síndrome del cromosoma X frágil". Tanto niños como niñas son susceptibles a padecerla, pero los niños se ven afectados con mayor frecuencia (tres veces más).
Epidemiología
El síndrome de Martin-Bell es una enfermedad bastante común: entre 0,3 y 1,0 de cada 1.000 hombres la padecen, y entre 0,2 y 0,6 de cada 1.000 mujeres. Además, los niños con síndrome de Martin-Bell nacen en todos los continentes con la misma frecuencia. Obviamente, la nacionalidad, el color de la piel, la forma de los ojos, las condiciones de vida y el bienestar de las personas no afectan la aparición de la enfermedad. Su frecuencia de aparición es comparable solo a la del síndrome de Down (1 enfermedad por cada 600-800 recién nacidos). Una quinta parte de los varones portadores del gen alterado son sanos, no presentan anomalías clínicas ni genéticas, el resto presenta signos de retraso mental, desde formas leves hasta graves. Entre las mujeres portadoras, algo más de un tercio están enfermas.
El síndrome del cromosoma X frágil afecta aproximadamente a 1 de cada 2500 a 4000 hombres y a 1 de cada 7000 a 8000 mujeres. Se estima que la prevalencia de portadores entre las mujeres es de 1 de cada 130 a 250, y entre los hombres, de 1 de cada 250 a 800.
Causas Síndrome de Martin-Bell
El síndrome de Martin-Bell se desarrolla debido al cese total o parcial de la producción corporal de una proteína específica. Esto ocurre debido a la falta de respuesta del gen FMR1, localizado en el cromosoma X. La mutación se produce como resultado de la reestructuración del gen a partir de variantes estructurales inestables de los estados génicos (alelos), y no desde el principio. La enfermedad se transmite únicamente por vía masculina, y el hombre no necesariamente padece la enfermedad. Los varones portadores transmiten el gen a sus hijas sin cambios, por lo que su retraso mental no es evidente. Con la transmisión posterior del gen de la madre a sus hijos, este muta y aparecen todos los signos característicos de esta enfermedad.
Patogenesia
La patogénesis del síndrome de Martin-Bell se basa en mutaciones del aparato genético, que provocan el bloqueo de la producción de la proteína FMR, una proteína vital para el organismo, especialmente en las neuronas, presente en diversos tejidos. Las investigaciones demuestran que las proteínas FMR participan directamente en los procesos de regulación de las traducciones que ocurren en el tejido cerebral. La ausencia de esta proteína o su producción limitada por el organismo provoca retraso mental.
En la patogenia de la enfermedad, la hipermetilación genética se considera un trastorno clave, pero aún no ha sido posible identificar definitivamente el mecanismo de desarrollo de este trastorno.
Al mismo tiempo, se descubrió la heterogeneidad del locus patológico, asociada tanto al polialelismo como al polilocus. Se determinó la presencia de variantes alélicas en el desarrollo de la enfermedad, causadas por la existencia de mutaciones puntuales y la destrucción del gen FMRL.
Los pacientes también presentan dos tripletes frágiles sensibles al ácido fólico, ubicados a 300 kb, así como a 1,5-2 millones de pb del triplete frágil que contiene el gen FMR1. El mecanismo de las mutaciones que se producen en los genes FRAXE y FRAXF (identificados en los tripletes frágiles mencionados anteriormente) está relacionado con el mecanismo de los trastornos del síndrome de Martin Bell. Este mecanismo se debe a la expansión de las repeticiones GCC y CGG, que provocan la metilación de las denominadas islas CpG. Además de la forma clásica de la patología, existen dos tipos raros que se diferencian por la expansión de las repeticiones de trinucleótidos (en la meiosis masculina y femenina).
Se descubrió que, en la forma clásica del síndrome, el paciente carece de una proteína nucleocitoplasmática especial del tipo FMR1, que se encarga de la unión de diversos ARNm. Además, esta proteína promueve la formación de un complejo que facilita los procesos de traducción dentro de los ribosomas.
Síntomas Síndrome de Martin-Bell
¿Cómo reconocer la enfermedad en niños? ¿Cuáles son los primeros signos? En los primeros meses de vida, es imposible reconocer el síndrome de Martin-Bell, salvo que a veces se observa una disminución del tono muscular. Después de un año, el cuadro clínico se hace más evidente: el niño empieza a caminar y hablar tarde, y en ocasiones el habla desaparece por completo. Es hiperactivo, mueve los brazos de forma aleatoria, le teme a las multitudes y al ruido, es terco, presenta arrebatos de ira, inestabilidad emocional, convulsiones epilépticas y no establece contacto visual. En pacientes con síndrome de Martin-Bell, la enfermedad también se delata por su apariencia: orejas prominentes y grandes, frente pesada, rostro alargado, mentón prominente, estrabismo, manos y pies anchos. También se caracterizan por trastornos endocrinos: a menudo sobrepeso, obesidad, testículos grandes en los hombres y pubertad precoz.

Entre los pacientes con síndrome de Martin-Bell, el nivel de inteligencia varía considerablemente: desde retraso mental leve hasta casos graves. Si una persona normal tiene un coeficiente intelectual (CI) promedio de 100 y un genio de 130, las personas susceptibles a la enfermedad tienen entre 35 y 70.
Todos los síntomas clínicos de la patología se pueden caracterizar por una tríada de manifestaciones principales:
- oligofrenia (CI 35-50);
- dismorfofobia (se observan orejas prominentes y prognatismo);
- macroorquidismo, que aparece después del inicio de la pubertad.
Aproximadamente el 80% de los pacientes también tienen prolapso de la válvula bicúspide.
Sin embargo, la forma completa del síndrome se manifiesta solo en el 60% de los pacientes. En el 10%, solo se detecta retraso mental, y en el resto, la enfermedad se presenta con una combinación diferente de síntomas.
Entre los primeros signos de la enfermedad, que aparecen a temprana edad:
- El niño enfermo presenta un retraso mental significativo en comparación con el desarrollo de otros compañeros;
- trastornos de atención y concentración;
- fuerte terquedad;
- Los niños empiezan a caminar y a hablar bastante tarde;
- Se observan hiperactividad y trastornos del desarrollo del habla;
- ataques de ira muy fuertes e incontrolables;
- Se puede desarrollar mutismo, es decir, una ausencia total del habla en el niño.
- El bebé experimenta ansiedad social y es capaz de entrar en pánico debido a ruidos fuertes o cualquier otro sonido fuerte;
- el niño agita los brazos de forma incontrolable y caótica;
- Se observa timidez, el niño tiene miedo de estar en lugares concurridos;
- la aparición de diversas ideas obsesivas, estado emocional inestable;
- Es posible que el bebé se muestre reacio a establecer contacto visual con las personas.
En los adultos se observan los siguientes síntomas de la patología:
- apariencia específica: una cara alargada con una frente pesada, orejas grandes y salientes, un mentón muy prominente;
- pie plano, otitis y estrabismo;
- la pubertad ocurre bastante temprano;
- puede desarrollarse obesidad;
- Con mucha frecuencia se observan defectos cardíacos en el síndrome de Martin-Bell;
- En los hombres se observa agrandamiento de los testículos;
- las articulaciones se vuelven muy móviles;
- El peso y la altura aumentan bruscamente.
Diagnostico Síndrome de Martin-Bell
Para diagnosticar el síndrome de Martin Bell, es necesario contactar con un genetista cualificado. El diagnóstico se realiza tras pruebas genéticas específicas que permiten identificar el cromosoma defectuoso.
Pruebas
En una etapa temprana de la enfermedad, se utiliza un método citogenético que consiste en extraer un fragmento de material celular del paciente y añadir ácido fólico para provocar cambios en los cromosomas. Tras un cierto tiempo, se identifica una zona del cromosoma con un adelgazamiento notable, lo que indica la presencia del síndrome del cromosoma X frágil.
Sin embargo, esta prueba no es adecuada para el diagnóstico en las últimas etapas de la enfermedad, porque su precisión se reduce por el uso generalizado de multivitaminas que contienen ácido fólico.
El diagnóstico integrado del síndrome de Martin-Bell es un examen genético molecular, que consiste en determinar el número de las llamadas repeticiones de trinucleótidos en el gen.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnóstico instrumental
Un método muy específico de diagnóstico instrumental es la PCR (reacción en cadena de la polimerasa), que permite estudiar la estructura de los residuos de aminoácidos contenidos en el cromosoma X y determinar así la presencia del síndrome de Martin Bell.
También existe un método independiente, aún más específico, para el diagnóstico patológico: una combinación de PCR y detección mediante electroforesis capilar. Este método es altamente preciso y detecta la patología cromosómica en pacientes con insuficiencia ovárica primaria, así como con síndrome atáxico.
La presencia del defecto puede determinarse mediante un electroencefalograma (EEG). Los pacientes con esta enfermedad presentan una actividad bioeléctrica cerebral similar.
¿Qué pruebas son necesarias?
Diagnóstico diferencial
Los métodos diferenciados que ayudan a sospechar el síndrome incluyen:
- clínico - el 97,5% de los pacientes tienen signos evidentes de retraso mental (moderado o profundo); el 62% tienen orejas grandes y prominentes; el 68,4% tienen un mentón y una frente grandes y prominentes; el 68,4% de los niños tienen testículos agrandados, el 41,4% tienen peculiaridades del habla (velocidad de habla desigual, volumen incontrolable, etc.);
- citogénico: se examina la sangre para el cultivo de linfocitos, se determina el número de células con cromosoma X frágil por cada 100 células estudiadas;
- Electroencefalografía: se registran los cambios en los impulsos eléctricos del cerebro que son específicos del síndrome de Martin-Bell.
¿A quién contactar?
Tratamiento Síndrome de Martin-Bell
En el tratamiento de pacientes adultos, se utilizan antidepresivos con psicoestimulantes. El tratamiento farmacológico es supervisado constantemente por un psicólogo y un psiquiatra. Además, en clínicas privadas se realizan microinyecciones con fármacos como Cerebrolysin (o sus derivados) y citomedinas (como Solcoseryl o Lidase).
En caso de desarrollo del síndrome atáxico, se utilizan anticoagulantes y nootrópicos. Además, se prescriben mezclas de aminoácidos y angioprotectores. A las mujeres con insuficiencia ovárica primaria se les prescribe un tratamiento correctivo con hierbas medicinales y estrógenos.
Los antagonistas del receptor de glutamina también se utilizan en el tratamiento.
Tradicionalmente, el tratamiento del síndrome de Martin-Bell consiste en el uso de medicamentos que inciden en los síntomas de la enfermedad, pero no en su causa. Esta terapia consiste en la prescripción de antidepresivos, neurolépticos y psicoestimulantes. No todos los fármacos están indicados para su uso en niños, por lo que la lista de fármacos es bastante limitada. Los neurolépticos que pueden utilizarse después de los 3 años (la edad más temprana para su prescripción) incluyen haloperidol en gotas y comprimidos, clorpromazina en solución y periciazina en gotas. Por lo tanto, la dosis de haloperidol para niños se calcula en función del peso corporal. Para los adultos, la dosis se prescribe individualmente. Se administra por vía oral, comenzando con 0,5-5 mg 2-3 veces al día, y luego se aumenta gradualmente a 10-15 mg. Cuando se observa mejoría, se reduce la dosis para mantener el estado alcanzado. En caso de agitación psicomotora, se prescriben 5-10 mg por vía intramuscular o intravenosa, con varias repeticiones tras 30-40 minutos. La dosis diaria no debe exceder los 100 mg. Es posible que se presenten efectos secundarios como náuseas, vómitos, espasmos musculares, aumento de la presión arterial, arritmia, etc. Las personas mayores deben tomar precauciones especiales, ya que se han registrado casos de paro cardíaco repentino y puede presentarse discinesia tardía (movimientos involuntarios).
Los antidepresivos aumentan la actividad de las estructuras cerebrales, alivian la depresión y la tensión, y mejoran el estado de ánimo. Estos fármacos, recomendados para el síndrome de Martin-Bell entre los 5 y los 8 años, incluyen clomipromina, sertralina, fluoxetina y fluvoxamina. Por lo tanto, la fluoxetina se toma por vía oral durante las comidas, 1 o 2 veces (preferiblemente en la primera mitad del día), comenzando con 20 mg al día y aumentando a 80 mg si es necesario. No se recomienda una dosis superior a 60 mg para las personas mayores. El tratamiento lo determina un médico, pero no debe durar más de 5 semanas.
Posibles efectos secundarios: mareos, ansiedad, tinnitus, pérdida de apetito, taquicardia, edemas, etc. Se requiere precaución al prescribir a ancianos, personas con enfermedades cardiovasculares y diabetes.
Los psicoestimulantes son fármacos psicotrópicos que se utilizan para mejorar la percepción de los estímulos externos: agudizan la audición, las reacciones de respuesta y la visión.
El diazepam se prescribe como sedante para neurosis, ansiedad, ataques epilépticos y convulsiones. Se administra por vía oral, intravenosa, intramuscular y rectal (en el recto). Se prescribe individualmente, según la gravedad de la enfermedad, con dosis mínimas de 5 a 10 mg y diarias de 5 a 20 mg. La duración del tratamiento es de 2 a 3 meses. En niños, la dosis se calcula teniendo en cuenta el peso corporal y las características individuales. Los efectos secundarios incluyen letargo, apatía, somnolencia, náuseas y estreñimiento. Es peligroso combinarlo con alcohol; puede causar adicción.
En el tratamiento del síndrome de Martin-Bell, se han observado casos de mejoría con la administración de fármacos de origen animal (cerebro): cerebrolisato, cerebrolisina y cerebrolisato-M. Los componentes principales de estos fármacos son péptidos que promueven la producción de proteínas en las neuronas, reponiendo así la proteína faltante. Cerebrolisina se administra en un chorro de 5 a 10 ml; el tratamiento consta de 20 a 30 inyecciones. El fármaco se prescribe a niños a partir de un año de edad, por vía intramuscular, 1-2 ml al día durante un mes. Es posible repetir la administración. Se han observado efectos secundarios como fiebre; está contraindicado en mujeres embarazadas.
Se intentó tratar la enfermedad con ácido fólico, pero solo mejoró el aspecto conductual (disminuyó la agresividad y la hiperactividad, mejoró el habla), y no se produjo ningún cambio a nivel intelectual. Para mejorar la condición de la enfermedad, se prescribe ácido fólico, se indican métodos de fisioterapia, logopedia y corrección pedagógica y social.
Los preparados de litio también se consideran eficaces, ya que ayudan a mejorar la adaptación del paciente al entorno social, así como su actividad cognitiva. Además, regulan su comportamiento en sociedad.
El uso de hierbas para el síndrome de Martin-Bell puede utilizarse como antidepresivo. Entre las hierbas que ayudan a aliviar la tensión, la ansiedad y a mejorar el sueño se encuentran la valeriana, la menta piperita, el tomillo, la hierba de San Juan y la manzanilla. Las infusiones se preparan de la siguiente manera: para una cucharadita de hierbas secas, se necesita un vaso de agua hirviendo. Las decocciones se dejan en infusión durante al menos 20 minutos, tomándose principalmente por la noche antes de acostarse o por la tarde. Una cucharada de miel sería un buen complemento.
Tratamiento de fisioterapia
Para eliminar las manifestaciones neurológicas, se realizan procedimientos fisioterapéuticos especiales, como ejercicios en la piscina, relajación muscular y acupuntura.
Tratamiento quirúrgico
Una etapa importante del tratamiento también se considera la cirugía plástica, que consiste en operaciones que ayudan a mejorar la apariencia del paciente. Se realiza cirugía plástica de las extremidades y las aurículas, así como de los genitales. También se realiza la corrección de la ginecomastia con epispadias, así como de otros defectos estéticos.
Prevención
El único método para prevenir la enfermedad es el cribado prenatal de las embarazadas. Existen exámenes especiales que permiten la detección temprana de la patología, tras lo cual se recomienda la interrupción del embarazo. Como alternativa, se utiliza la FIV, que puede ayudar al niño a heredar un cromosoma X sano.
La prevención del paciente depende de si la mutación genética ha reaparecido o ha sido hereditaria. Para ello, se realiza un diagnóstico genético molecular. El hecho de que la prueba no haya revelado el "cromosoma X frágil" en familiares indica que la mutación es reciente, lo que significa que el riesgo de tener un hijo con síndrome de Martin-Bell es muy bajo. En familias con personas enfermas, la prueba ayudará a evitar la recurrencia de casos.
Pronóstico
El pronóstico del síndrome de Martin-Bell es favorable para la vida, pero no para la recuperación. La esperanza de vida depende de la gravedad de la enfermedad y los defectos asociados. El paciente puede llevar una vida normal. En las formas graves del síndrome de Martin-Bell, los pacientes corren el riesgo de sufrir una discapacidad de por vida.
Esperanza de vida
El síndrome de Martin Bell no tiene un impacto negativo grave en la salud, por lo que la esperanza de vida de la mayoría de las personas a las que se les ha diagnosticado esta patología no difiere de los indicadores estándar.