
Todo el contenido de iLive se revisa médicamente o se verifica para asegurar la mayor precisión posible.
Tenemos pautas de abastecimiento estrictas y solo estamos vinculados a sitios de medios acreditados, instituciones de investigación académica y, siempre que sea posible, estudios con revisión médica. Tenga en cuenta que los números entre paréntesis ([1], [2], etc.) son enlaces a estos estudios en los que se puede hacer clic.
Si considera que alguno de nuestros contenidos es incorrecto, está desactualizado o es cuestionable, selecciónelo y presione Ctrl + Intro.
Lisencefalia cerebral
Médico experto del artículo.

Último revisado: 12.07.2025
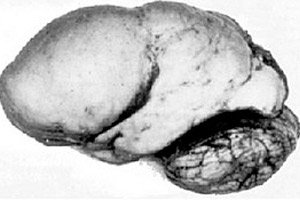
Entre las patologías cerebrales orgánicas se destaca una anomalía congénita del desarrollo cerebral llamada lisencefalia, cuya esencia radica en la superficie casi lisa de la corteza de sus hemisferios, con un número insuficiente de circunvoluciones y surcos. [ 1 ]
En ausencia total de circunvoluciones, se define agiria, y la presencia de varias circunvoluciones anchas y planas se denomina paquigiria. Estos defectos, al igual que otras deformaciones por reducción del cerebro, se clasifican como Q04.3 en la CIE-10.
Epidemiología
Según las estadísticas sobre enfermedades raras, hay entre 1 y 1,2 casos de lisencefalia por cada 100 mil recién nacidos. [ 2 ], [ 3 ]
Según algunos datos, en niños con síndrome de Miller-Dieker se observan hasta un 25-30% de casos de lisencefalia clásica; en casi el 85% de los pacientes se detectan mutaciones puntuales y deleciones de los genes LIS1 y DCX. [ 4 ]
Los estudios genéticos de 17 genes asociados con la lisencefalia mostraron que la mutación o deleción de LIS1 representa el 40% de los pacientes, y el 23% está asociado con la mutación DCX, seguida de TUBA1A (5%) y DYNC1H1 (3%).[ 5 ]
Causas lisencefalia
Todas las causas conocidas de la formación de la corteza cerebral (cortex cerebri), casi o completamente sin circunvoluciones ni surcos, que aumentan el área de trabajo del cerebro humano y aseguran la productividad del sistema nervioso central, se asocian con alteraciones en su desarrollo perinatal. Es decir, se desarrolla lisencefalia en el feto. [ 6 ]
La falla en la formación de las capas de la corteza cerebral del feto en la lisencefalia es el resultado de una migración anormal de las neuronas que la forman o del cese prematuro de este proceso.
Este proceso, esencial para la histogénesis cerebrocortical, ocurre en varias etapas entre la semana 7 y la 18 del embarazo. Dada su mayor sensibilidad a las mutaciones genéticas, así como a diversas influencias físicas, químicas y biológicas negativas, cualquier desviación de la norma puede provocar una localización incorrecta de las neuronas, con la posible formación de una capa engrosada de materia gris de la corteza sin una estructura característica. [ 7 ]
En algunos casos, la lisencefalia en niños se asocia con los síndromes de Miller-Dieker, Walker-Warburg o Norman-Roberts.
Lea también: Defectos del desarrollo cerebral
Factores de riesgo
Además de las mutaciones en algunos genes, los factores de riesgo para el nacimiento de un niño con un defecto tan grave incluyen la falta de oxígeno (hipoxia) del feto; suministro insuficiente de sangre al cerebro (hipoperfusión); accidente cerebrovascular agudo en forma de accidente cerebrovascular perinatal; patologías placentarias; infecciones virales de la mujer embarazada (incluido TORCH); [ 8 ] problemas con el metabolismo general y la función tiroidea; tabaquismo, alcohol, sustancias psicotrópicas y narcóticas; uso de varios medicamentos; niveles elevados de radiación. [ 9 ]
Patogenesia
No todos los casos de lisencefalia tienen una patogénesis causada por anomalías cromosómicas y mutaciones genéticas. Sin embargo, se conocen algunos genes que codifican proteínas que desempeñan un papel importante en el correcto movimiento de neuroblastos y neuronas a lo largo de las células gliales radiales para formar la corteza cerebral. Las mutaciones de estos genes conducen a esta patología. [ 10 ]
En particular, se trata de mutaciones esporádicas (sin herencia) del gen LIS1 en el cromosoma 17, que regula la proteína motora citoplasmática de los microtúbulos, la dineína, así como del gen DCX en el cromosoma X, que codifica la proteína doblecortina (lisencefalina-X). [ 11 ] En el primer caso, los especialistas definen la lisencefalia clásica (tipo I) y, en el segundo, la ligada al cromosoma X. [ 12 ]
Cuando se elimina el gen FLN1, que codifica la fosfoproteína filamina 1, el proceso de migración neuronal dirigida puede no comenzar en absoluto, lo que lleva a una ausencia total de convoluciones (agiria). [ 13 ]
Se han identificado mutaciones en el gen CDK5, que codifica una enzima quinasa, un catalizador del metabolismo intracelular que regula el ciclo celular en las neuronas del SNC y asegura su migración normal durante la formación prenatal de las estructuras cerebrales.
Los cambios anormales en el gen RELN en el cromosoma 7 que causan defectos en la circunvolución cortical en el síndrome de Norman-Roberts resultan en una deficiencia de la glucoproteína extracelular reelina, que es necesaria para regular la migración y el posicionamiento de las células madre neurales durante el desarrollo de la corteza cerebral.[ 14 ], [ 15 ], [ 16 ]
El gen ARX codifica la proteína homeobox no aristalens, un factor de transcripción que desempeña un papel importante en el prosencéfalo y otros tejidos.[ 17 ] Los niños con la mutación ARX tienen otros síntomas, como partes faltantes del cerebro (agenesia del cuerpo calloso), genitales anormales y epilepsia grave.[ 18 ],[ 19 ]
Varios genes se han vinculado a la lisencefalia, entre ellos VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A y CDK5.[ 20 ]
El citomegalovirus (CMV) se ha asociado con el desarrollo de lisencefalia debido a la disminución del riego sanguíneo al cerebro fetal. La gravedad de la infección por CMV depende de la edad gestacional. Es más probable que la infección temprana cause lisencefalia debido a que la migración neuronal ocurre al inicio del embarazo.[ 21 ]
Además, el mecanismo de aparición de esta anomalía incluye el cese incompleto o tardío de la migración neuronal desde la zona generativa periventricular a la corteza cerebral. En tales casos, se desarrolla lisencefalia incompleta o paquigiria, en la que se forman varios surcos y circunvoluciones amplios (aunque la mayoría están ausentes).
Síntomas lisencefalia
Los primeros signos de esta patología (en ausencia de los síndromes mencionados anteriormente) pueden no aparecer inmediatamente después del nacimiento, sino después de un mes y medio o dos meses. Con mayor frecuencia, se observan los siguientes síntomas clínicos de lisencefalia:
- hipotonía muscular, a menudo combinada con parálisis espástica;
- convulsiones y crisis tónico-clónicas generalizadas (en forma de opistótonos);
- retraso mental grave y retraso del crecimiento;
- deterioro de las funciones neurológicas y motoras.
Los problemas con la deglución provocan dificultades a la hora de alimentar al bebé. [ 22 ]
Un alto grado de deterioro neuromotor a menudo se manifiesta como tetraplejía (parálisis de todas las extremidades). Es posible la deformación de las manos, los dedos de las manos o de los pies.
En el síndrome de Norman-Roberts con lisencefalia tipo I se observan anomalías craneofaciales: microcefalia severa, frente baja y puente nasal ancho y prominente, ojos muy separados (hiperplasia prostática benigna), subdesarrollo de las mandíbulas (micrognatia). [ 23 ]
El síndrome de Miller-Dieker también puede caracterizarse por un tamaño de cabeza anormalmente pequeño con una frente ancha y alta y una nariz corta, depresiones en las sienes (depresiones bitemporales) y orejas deformadas y de implantación baja.
El síndrome de lisencefalia grave se caracteriza por microcefalia, una disminución del tamaño de los globos oculares (microftalmia), combinada con displasia de retina, hidrocefalia obstructiva y cuerpo calloso ausente o hipoplásico.
Complicaciones y consecuencias
Entre las complicaciones de esta anomalía, los especialistas mencionan la violación de la función de deglución (disfagia) y el reflujo gastroesofágico; epilepsia refractaria (no controlada); infecciones frecuentes del tracto respiratorio superior; neumonía (incluida la aspiración crónica).
Los bebés con lisencefalia pueden tener problemas cardíacos orgánicos congénitos en forma de un defecto del tabique auricular o un defecto cardíaco complejo con cianosis (tetralogía de Fallot). [ 24 ]
Las consecuencias del retraso del crecimiento postnatal en la mayoría de los casos resultan en la muerte dentro de los 24 meses posteriores al nacimiento.
Diagnostico lisencefalia
El diagnóstico comienza con un examen físico del niño, un estudio de la historia clínica de los padres y la historia del embarazo y el parto.
Durante la gestación, puede ser necesario realizar pruebas de ADN fetal libre de células, amniocentesis o muestreo de vellosidades coriónicas. [ 25 ] Para obtener más información, consulte – Diagnóstico prenatal de enfermedades congénitas
Los diagnósticos instrumentales se utilizan para visualizar las estructuras cerebrales y evaluar sus funciones:
- tomografía computarizada del cerebro;
- resonancia magnética (MRI) del cerebro;
- electroencefalograma (EEG). [ 26 ]
Durante el embarazo, se puede sospechar lisencefalia en la ecografía del feto después de las 20-21 semanas por la ausencia de los surcos parietooccipital y calcarino y una anomalía de la cisura de Silvio del cerebro.
Diagnóstico diferencial
Se realiza diagnóstico diferencial con otros síndromes de defectos cerebrales congénitos.
Existen más de 20 tipos de lisencefalia, la mayoría de los cuales se dividen en dos categorías principales: lisencefalia clásica (tipo 1) y lisencefalia en empedrado (tipo 2). Cada categoría presenta manifestaciones clínicas similares, pero diferentes mutaciones genéticas.[ 27 ]
El examen cerebral en la lisencefalia tipo I muestra una corteza cerebral con cuatro capas en lugar de las seis que se observan en pacientes normales, mientras que en la lisencefalia tipo 2 la corteza cerebral está desorganizada y presenta un aspecto abultado o nodular debido al desplazamiento completo de la corteza cerebral por grupos de neuronas corticales separados por tejido gliomesenquimal. Los pacientes también presentaban anomalías musculares y oculares.
- Lisencefalia clásica (tipo 1):
- LIS1: Lisencefalia aislada y síndrome de Miller-Dieker (lisencefalia asociada con dismorfia facial). [ 28 ]
- LISX1: Mutación del gen DCX. En comparación con la lisencefalia causada por mutaciones en LIS1, el DCX presenta una corteza de seis capas en lugar de cuatro.
- Lisencefalia aislada sin otros defectos genéticos conocidos
- Lisencefalia en adoquín (tipo 2):
- Síndrome de Walker-Warburg
- Síndrome de Fukuyama
- Enfermedades de los músculos, ojos y cerebro.
- Los demás tipos no pueden incluirse en ninguno de los dos grupos anteriores:
- LIS2: Síndrome de Norman-Roberts, similar a la lisencefalia tipo I o síndrome de Miller-Dieker, pero sin la deleción del cromosoma 17.
- LIS3
- LISX2
Microlisencefalia: Se trata de una combinación de ausencia de plegamiento cortical normal y una cabeza anormalmente pequeña. Los niños con lisencefalia regular tienen una cabeza de tamaño normal al nacer. A los niños con una cabeza pequeña al nacer se les suele diagnosticar microlisencefalia.
También es importante distinguir entre lisencefalia y polimicrogiria, que son diferentes defectos del desarrollo cerebral.
¿A quién contactar?
Tratamiento lisencefalia
La lisencefalia es un defecto orgánico incurable, por lo que sólo es posible un tratamiento de apoyo y sintomático. [ 29 ]
En primer lugar, se trata del uso de anticonvulsivos y antiepilépticos, así como de la colocación de una sonda de gastrostomía (si el niño no puede tragar por sí solo). El masaje es útil.
En casos de hidrocefalia grave, se extrae líquido cefalorraquídeo.
Prevención
Los expertos recomiendan que los futuros padres busquen asesoramiento genético y que las mujeres embarazadas se registren a tiempo con los obstetras y ginecólogos y se sometan a todos los exámenes programados.
Pronóstico
En los niños con lisencefalia, el pronóstico depende de su grado, pero en la mayoría de los casos, el desarrollo mental del niño no supera los cuatro o cinco meses. Todos los niños con este diagnóstico padecen trastornos psicomotores graves y epilepsia de difícil tratamiento. [ 30 ]
Según el NINDS (Instituto Nacional Estadounidense de Trastornos Neurológicos y Accidentes Cerebrovasculares), la esperanza de vida máxima para las personas con lisencefalia es de unos 10 años.